Glossaire
Essais cliniques - Région Sud Méditerranée
Séminaire Ketty Schwartz
Ce glossaire est destiné à toute personne qui s'intéresse voire s'implique dans la recherche clinique.
Délibérément, certaines définitions ont été simplifiées par rapport à celles, plus "orthodoxes" rencontrées dans des ouvrages techniques.
Au-delà d'une définition assez brève, la plupart des articles comporte un complément destiné à resituer le terme dans son contexte. Dans un même esprit, d'autres termes proches ou opposés sont mentionnés comme des invitations à circuler à l'intérieur de ce document.
Certaines définitions sont inspirées du travail du réseau "EUPATI", d'autres sont issues de la collaboration avec OrphanDev, F-Crin, et l'AFA à l'occasion de la formation "Explique-moi les essais cliniques". Qu'ils en soient tous remerciés ici. Nous souhaitons également mentionner le" Dictionnaire de pharmaco-épidémiologie" de Bernard Bégaud (ARME-Pharmacovigilance Editions) consulté en préparant cette formation.
Nous vous proposons également, en fin de document le décryptage de quelques sigles et acronymes fréquemment rencontrés dans ce domaine.
Enfin, nous avons souhaité inclure quelques graphismes d'Alexandra Pinci, comme autant de clins d'œil au séminaire que nous avons partagé, dans un document par nature assez austère.
Marion Mathieu "Tous Chercheurs"
François Faurisson Inserm
François Faurisson Inserm

Autorisation de Mises sur le Marché
Agence Nationale de Sécurité du Médicament et des Produits de Santé
Assistant de Recherche Clinique
Amélioration du Service Médical Rendu
Autorisation temporaire d'utilisation
Bonnes Pratiques Cliniques
Comité Consultatif National d'Ethique
Comité d'Ethique de l'Inserm
Comité d’Evaluation Ethique de l'Inserm
Comité d'Ethique de la Recherche
Centre Hospitalo-Universitaire
Centre d’investigation Clinique
Commission Nationale Informatique et Liberté
Comité de Protection des Personnes
Centre de Ressources Biologiques
Case Report Form : Cahier d'observation
Contract Research Organisation : Organisme prestataire de services en recherche
Code de la Santé Publique
Dénomination Commune Internationale
Dispositifs Médicaux
Data Safety Monitoring Board : Comité de surveillance indépendant
European Medicine Agency : Agence Européenne du Médicament
Haute Autorité de Santé
International Conference on Harmonisation : Conférence internationale d'harmonisation
Institut National de la Santé et de la Recherche Médicale
Institutional Review Board : Comité d'éthique de la recherche
Intention to treat : Intention de traiter
Notice d'Information du Formulaire de Consentement
No Observable Adverse Effect Level : Seuil « aucun effet indésirable observé »
Pharmacodynamie
Pharmacokinetics : Pharmacocinétique
Qualité de Vie
Résumé des caractéristiques du produit
Service Médical Rendu
-
A
-
B
-
C
-
D
-
E
-
F
-
G
-
H
-
I
-
J
-
L
-
M
-
N
-
O
-
P
-
Q
-
R
-
S
-
T
-
U
-
V
Absorption (Pharmacologie)
Passage d’un médicament de son site d’administration à la circulation sanguine.
L’absorption est caractérisée par la proportion de médicament atteignant le plasma, appelée biodisponibilité exprimée en pourcentage de la dose administrée, et la vitesse à laquelle se produit ce phénomène. Pour un même principe actif, l’absorption varie selon la voie d’administration (orale, cutanée, intraveineuse,…). La voie intraveineuse a, par définition, une biodisponibilité de 100%, et un présence immédiate dans la circulation sanguine.
-> Distribution Elimination, Métabolisme, Pharmacocinétique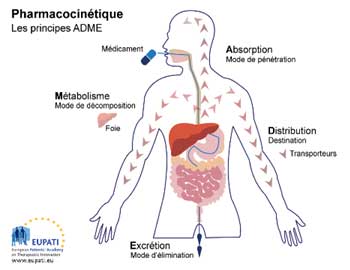
Admissibilité (Méthodologie)
voir : Eligibilité
-> Critères d’inclusion, Eligibilité
Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) (Règlementation)
Organisme public dépendant du ministère de la Santé responsable de la mise sur le marché et de la surveillance d’utilisation des produits de santé.
L’ANSM délivre également les autorisations de recherche clinique menées sur le territoire. L’ANSM s’est substituée le 1er mai 2012 à l’Agence française de sécurité sanitaire du médicament et des produits de santé (Afssaps) dont elle a repris les missions, droits et obligations. Elle a été dotée de responsabilités et de missions nouvelles, de pouvoirs et de moyens renforcés. Il s’agit d’une structure nationale ; à l’échelle Européenne, l’EMA (Agence Européenne du Médicament) a également des missions de contrôle de mise sur le marché et de surveillance d’utilisation de produits de santé. (pour d’infos : http: //ansm.sante.fr/)
-> Autorisation de Mise sur le Marché, European Medicines Agency, Pharmacovigilance
Amélioration du service médical rendu (ASMR) (Règlementation)
Evaluation comparative du Service Médical Rendu avec les traitements déjà existants.
Déterminée par la commission de transparence de la Haute Autorité de Santé (HAS) l’ASMR, chiffrée de 0 : apport extrêmement innovant dans le traitement de la maladie à 5, sans avantage par rapport aux traitements existants, l’ASMR est un élément clé de la fixation de son prix après obtention de l’Autorisation de Mise sur le Marché.
-> Autorisation de Mise sur le Marché, SMR
Analyse « per protocole » (Méthodologie)
Analyse des résultats d’une recherche clinique ne tenant compte que des participants ayant suivi l’ensemble du traitement expérimental.
Ce mode d’analyse favorise la mise en évidence des propriétés pharmacodynamiques d’un médicament, mais s’éloigne des conditions normales d’utilisation, notamment en occultant les arrêts de traitement liés à des effets indésirables ou des contraintes du traitement. L’analyse « per protocole » s’oppose à l’analyse « en intention de traiter ». Cette option d’analyse doit être prévue dans le protocole.
-> Analyse en intention de traiter Observance
Analyse en intention de traiter (ITT) (Méthodologie)
Analyse des résultats d’une recherche clinique de tous les participants sans prendre en compte l’observance des participants.
Ce mode d’analyse se rapproche de conditions normales d’utilisation d’un médicament, en évitant par exemple de conclure à l’efficacité d’un médicament dont seule une minorité de participants « observants » seraient évalués, les autres l’ayant interrompu en raison d’effets indésirables fréquents. L’analyse « en intention de traiter » s’oppose à l’analyse « per protocole ». Cette option d’analyse doit être prévue dans le protocole. Terme anglais : intention to treat.
-> Observance, Analyse per protocole
Analyse intermédiaire (Méthodologie)
Toute analyse destinée à comparer l'efficacité ou l'innocuité du traitement des différents groupes de traitement, à n'importe quel moment avant la fin formelle d'un essai clinique.
Une analyse intermédiaire doit être incluse dans le plan d'analyse statistique, qui fait partie du protocole. Elle peut conduire à interrompre l’essai, soit parce que la réponse à la question posée est déjà obtenue, soit parce que dans les conditions prévues par le protocole aucune réponse ne pourra être obtenue (essai non conclusif). L’analyse intermédiaire, programmée, est à distinguer d’une levée d’aveugle prescrite en situation d’urgence par le comité de surveillance de l’étude, généralement à la suite d’évènements indésirables graves.
-> Méthodes adaptatives, Comité indépendant de surveillance
Anonymisation (Ethique)
Suppression des informations personnelles (telles que les noms et coordonnées des participants à un essai clinique) de la base de données de l’essai, interdisant l’identification des individus y participant.
Une véritable anonymisation des données implique l’absence de données identifiantes qui, notamment dans le cas de maladies ou de situations rares, pourraient permettre par recoupements, l’identification d’individus. Par exemple une femme dont on sait qu’elle est née en 1964, mère de 7 enfants et habite dans un département donné risque d’être identifiable sans que son identité apparaisse explicitement. Dans de telles situations le respect de la confidentialité demande de ne pas communiquer de données individuelles, mais les données dites « agrégées » de différents individus.
-> Confidentialité, Respect de la personne
Arrêt anticipé d'un essai clinique (Règlementation)
Arrêt prématuré d'un essai clinique, quel qu'en soit le motif, avant que les conditions indiquées dans le protocole ne soient remplies.
-> Analyse intermédiaire Comité indépendant de surveillance
Arrêt temporaire d'un essai clinique (Règlementation)
Interruption non prévue par le protocole de la conduite d'un essai clinique par le promoteur dans l'intention de le reprendre.
-> Comité indépendant de surveillance
Assistant ou attaché de recherche clinique (ARC) (Méthodologie)
Personne qualifiée mandatée par le promoteur et chargée d’assurer la qualité d’une recherche portant sur la personne humaine.
Dans un essai clinique, il doit veiller au respect des droits des patients, du protocole de l’étude, des Bonnes Pratiques Cliniques et des procédures opératoires standard de l’essai.
-> Investigateur, Promoteur
Assurance de l’essai clinique (Règlementation)
Assurance en responsabilité civile couvrant les éventuels dommages subis par les patients ou sujets sains participant à l'essai.
Avant le début d'un essai clinique, le promoteur doit avoir contracté cette assurance. Il s'agit d'une assurance de type "responsabilité pour faute", c'est à dire que le patient (plaignant) doit être indemnisé en cas de dommage sauf si l'assureur arrive à démontrer qu'il n'existe aucun lien de cause à effet entre le dommage et la recherche à laquelle a participé le patient ou le sujet sain. Toute réclamation d'un patient s'estimant victime d'un dommage lié à une recherche portant sur la personne humaine doit être effectuée dans les 10 ans après la fin de la recherche. (article L1121-10 du Code de Santé Publique)
-> Promoteur
Assurance qualité (Méthodologie)
Ensemble des activités préétablies et systématiques mises en œuvre pour s’assurer que la recherche est réalisée conformément aux bonnes pratiques cliniques et aux dispositions législatives et règlementaires en vigueur
L’assurance qualité inclut la manière dont les données sont générées, recueillies par écrit, documentées, enregistrées et rapportées.
-> Bonnes pratiques cliniques
Autonomie (Ethique)
Fait de se donner à soi-même sa propre loi.
L’autonomie est la notion fondamentale de la morale de Kant. C’est seulement lorsque l’individu s’impose à lui-même une loi qu’il agit moralement. En outre, il réalise ainsi sa liberté, qui ne peut se réaliser dans le vide, mais seulement par “l’intermédiaire de la loi”.
-> Ethique, Respect de la personne
Autorisation de mise sur le marché (AMM) (Règlementation)
Autorisation administrative de commercialisation d’un médicament délivrée sur avis de l’Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) à l'établissement pharmaceutique le produisant.
Cet avis tient compte de trois éléments : - l'efficacité - la sécurité - la qualité pharmaceutique de fabrication du médicament. Depuis la création de l’EMA (Agence Européenne du Médicament), l’AMM peut être centralisée, c’est-à-dire délivrée, après avis des agences nationales, pour tous les pays de l’Union Européenne.
-> Agence Nationale de Sécurité du Médicament et des Produits de Santé, European Medicine Agency, Soumission
Autorisation temporaire d'utilisation (ATU) (Règlementation)
Procédure de l'ANSM permettant l'utilisation d'un médicament n'ayant pas encore d’AMM.
L'ATU peut être accordée à un malade (ATU nominative délivrée à la demande du médecin traitant) ou à un groupe de malades atteints de la même maladie (ATU de cohorte dont l'organisation revient au laboratoire pharmaceutique titulaire de l'autorisation temporaire). L'ATU fait partie des modes d'accès dits compassionnels au médicament. Elle précède en règle une AMM mais n’est pas associée à une procédure d’évaluation propre : L'ATU ne vise pas à améliorer les connaissances médicales mais à mettre un médicament à disposition des malades dans des conditions exceptionnelles.
-> Autorisation de Mise sur le Marché
Aveugle (Méthodologie)
voir : Insu
Base de données (Méthodologie)
Ensemble d’informations recueillies et organisées systématiquement en vue d’une analyse.
Dans le cadre d’un essai clinique, on dit que la "base de l’essai est gelée" lorsqu’il n’est plus possible de modifier le contenu de la base de données. A ce moment, les études statistiques peuvent débuter sur ces données définitives.
-> Commission Nationale Informatique et Liberté, Confidentialité, Saisie.
Bénéfice thérapeutique (Méthodologie)
Amélioration de l’état de santé ou du bien-être d’une population de patients soumis à un traitement ou une stratégie thérapeutique.
On distingue le bénéfice en termes de morbidité (diminution des complications de la maladie ou amélioration de l’état de santé) et le bénéfice en termes de mortalité (diminution du nombre de décès observés dans un groupe de patients) pendant une période déterminée.
-> Morbidité, Mortalité, Santé publique
Biais (Méthodologie)
Erreur systématique dans la conduite ou l’analyse d’une expérimentation.
Le biais peut être « opérationnel », lorsqu’il est dû à la façon dont est conduit un essai (inclusion des participants, administration du produit, recueil des critères d’évaluation) ou « statistique » lorsqu’il est dû à la façon dont les résultats sont analysés ou évalués. Ainsi dans un essai en ouvert ou en simple insu, la connaissance du traitement peut conduire l’investigateur, consciemment ou non, à se comporter différemment avec les participants selon leur groupe de traitement (biais d'évaluation). L’exclusion de données de certains participants pour lesquels les résultats sont connus constitue également un biais voire une fraude lorsqu’elle est intentionnelle. Les meilleures techniques pour concevoir une méthodologie d’essai clinique qui évite les biais sont la randomisation et l’insu maintenu jusqu’à l’analyse des résultats. L’effet potentiel d’un biais doit toujours être recherché et pris en compte lors de l’analyse statistique des données de l’essai.
-> Randomisation
Biais d’inclusion (Méthodologie)
Déséquilibre entre les groupes de traitement lors de l’inclusion concernant une caractéristique individuelle pouvant influer sur l’effet du traitement (âge, stade de la maladie, etc.).
La randomisation dans l’attribution des traitements permet d’éviter les biais d’inclusion.
-> Randomisation
Bienfaisance (Ethique)
Un des concepts majeurs de l'éthique hippocratique de la médecine et de la recherche clinique.
Il stipule que l’intérêt et le bien être des participants doivent être pris en compte à chaque étape de la recherche. Les risques inhérents à toute recherche clinique ont fait préférer l’expression «non malfaisance » à « bienfaisance ». C’est le principe de bienfaisance qui interdit l’utilisation d’un placebo comme comparateur, si un traitement de référence existe. L’utilisation du placebo à la place du traitement de référence en diminuant les chances d’amélioration des participants concernés serait malfaisante.
-> Autonomie, Comparateur Ethique, Justice distributive, Non malfaisance, Respect de la personne
Biobanque (Méthodologie)
Vaste recueil organisé d’échantillons, habituellement humains, utilisés pour la recherche.
Les biobanques répertorient et stockent des échantillons selon des caractéristiques génétiques, cliniques et autres (âge, sexe, groupe sanguin et ethnicité par exemple). Certains échantillons sont aussi catégorisés en fonction des facteurs environnementaux, tels que l’exposition du donneur à certaines substances. Les biobanques jouent un rôle capital dans la recherche portant sur la personne humaine, comme en génomique et en médecine personnalisée. Les chercheurs s’adressent aux biobanques lorsqu’ils ont besoin d’échantillons avec des caractéristiques précises pour leurs travaux.
-> EuroBioBank, Saisie, Commission Nationale Informatique et Liberté
Biomarqueur (Méthodologie)
Mesure biologique étroitement associée à une caractéristique de la maladie d’un individu donné à un moment donné.
Un biomarqueur révélant l’existence d’une maladie a une valeur diagnostique. S’il est associé à l’évolution prévisible de la maladie, il s’agit d’un biomarqueur pronostique. Il peut s’agir également de mesures électrophysiologiques (espace entre les ondes Q et T dans l’électrocardiogramme pour les effets indésirables cardiaques de certains médicament) ou d’imagerie (IRM cérébrale dans l’évolution de la sclérose en plaque). Les biomarqueurs sont utilisés dans de nombreux domaines scientifiques, de différentes manières et à différents stades du développement des médicaments, y compris, dans certains cas, comme critère d’évaluation de substitution pour indiquer et mesurer l’effet des médicaments dans les essais cliniques. Par exemple, le taux d’hémoglobine est utilisé dans les essais de phase III dans le cadre du développement de traitements de la maladie de Gaucher de type 1. Cette maladie rare touche de multiples systèmes d’organes et raccourcit l’espérance de vie, mais il faut parfois des années avant que la maladie n’évolue sur le plan clinique. Par conséquent, l’évolution clinique n’est pas un bon critère d’évaluation de l’impact de nouveaux traitements sur cette maladie et il est utile de disposer de biomarqueurs qui permettent de déceler plus tôt les changements souhaités.
-> Critère d’évaluation, Critère d’évaluation de substitution
Biomédicament (Pharmacologie)
voir : Médicament biologique
Bonnes Pratiques Cliniques (BPC) (Méthodologie)
Ensemble de dispositions ayant pour but de concourir à la protection des droits, à la sécurité et à la protection des personnes se prêtant à ces recherches ainsi qu’à la crédibilité et à la confidentialité des données à caractère personnel et des résultats de ces recherches.
Le premier texte français des BPC date de 1987. Par la suite, les BPC ont été harmonisées entre l’Europe, les USA et le Japon dans le cadre des conférences internationales d’harmonisation (ICH).
Bras d’essai (Méthodologie)
voir : Groupe de traitement
-> Essai comparatif, Comparateur, Randomisation
Cahier d’observation (Méthodologie)
Support papier ou électronique qui contient l’ensemble des informations individuelles à recueillir lors de l’essai clinique qui devront être analysées ultérieurement.
Ce document est rempli par le médecin investigateur d’un essai ou un technicien/assistant de recherche clinique. Comme le dossier médical d’une personne, sa confidentialité doit être protégée Termes anglais : Case Report Form ou CRF (et electronic Case Report Form ou eCRF pour sa version électronique).
-> Saisie
Causalité (Méthodologie)
Lien établi entre une cause et un effet.
Un lien de causalité est requis pour établir la responsabilité d’un médicament dans un effet indésirable.
-> Effet indésirable, Evènement indésirable
Centre de ressource biologiques (CRB) (Méthodologie)
voir : Biobanque
Centre d'investigation clinique (CIC) (Méthodologie)
Infrastructure de recherche clinique mises à la disposition des investigateurs pour y réaliser leurs projets de recherche clinique et en santé.
Ils sont gouvernés par une double tutelle, la Direction générale de l’offre de soins (DGOS) du ministère de la Santé et des Sports et l’Inserm. Les CIC incluent des modules "Épidémiologie clinique", structures méthodologiques de soutien à la recherche clinique et épidémiologique, des modules "Innovations technologiques", structures dédiées à l’évaluation et à l’innovation de biomatériaux, de dispositifs médicaux, de logiciels consacrés à la santé, et des modules "Intégrés en Biothérapies" qui réalisent des projets de recherche innovants notamment en thérapie cellulaire et génique, immunothérapie, et vaccination.
Clinicaltrials.gov (Méthodologie)
Base de données en ligne qui fournit des informations sur des études cliniques sur une large gamme de maladies.
On y trouve des informations indiquant si les études recrutent des patients et puis un résumé des résultats des études une fois qu'elles sont terminées. La base de données est disponible à l'adresse suivante : https: //clinicaltrials.gov (adresse correcte en mai 2017). Il est possible de rechercher les études en fonction d’une maladie ou d’un pays. Bien que fondée par le gouvernement des États-Unis, cette ressource inclut des études basées dans le monde entier. Ce site Web s'adresse aux patients et à leurs familles, aux professionnels de santé, aux chercheurs et au public. Il est tenu à jour par la National Library of Medicine (NLM) aux National Institutes of Health (NIH).
Code de Nüremberg (Ethique)
Liste de dix critères, contenue dans le jugement du procès des médecins de Nüremberg en 1947.
Le Code de Nuremberg identifie le consentement éclairé comme préalable absolu à la conduite de recherche mettant en jeu des sujets humains.
-> Ethique, Respect de la personne, Bienfaisance
Code de la Santé Publique (CSP) (Règlementation)
Ensemble des textes législatifs qui régissent les questions de la santé publique en France.
Il est le garant de la déontologie médicale. Ses articles L1121 à L1125 portent sur la recherche clinique.
-> Déontologie
Comité Consultatif National d'Ethique (CCNE) (Ethique)
Comité chargé de donner des avis sur les problèmes éthiques et les questions de société soulevés par les progrès de la connaissance dans les domaines de la biologie, de la médecine et de la santé.
Composé de 39 membres et d'un président, sa composition est multidisciplinaire. Ses membres sont nommés pour part par le Président de la république, divers ministères, organismes de recherche, et universités.
-> Ethique. Comité d'éthique de la recherche
Comité d'Ethique de l'Inserm (CEI) (Ethique)
Comité ayant pour mission d’animer la réflexion sur les questions éthiques soulevées par la recherche scientifique médicale et la recherche en santé telle qu’elle sont mises en œuvre au sein de l’Institut.
-> Ethique. Comité d'éthique de la recherche
Comité d'Ethique de la Recherche (CER) (Ethique)
Comité multidisciplinaire qui veille à assurer la sécurité et le bien-être des participants aux projets de recherche.
Il s’assure que la recherche se déroule conformément aux principes scientifiques et éthiques. L'enregistrement d'une recherche clinique auprès d'un CER est requis lors de la publication des résultats dans de nombreux journaux scientifiques. Terme anglais : Institutional Review Board.
-> Ethique. Comité d'éthique de l'Inserm
Comité d'évaluation éthique de l'Inserm (CEEI) (Ethique)
Comité qui rend des avis sur des projets de recherche portés en priorité par des chercheurs de l'Inserm et du CNRS.
Le CEEI est un comité d'éthique de la recherche (CER) tel que défini par le Conseil de l'Europe.
-> Ethique. Comité d'éthique de la recherche
Comité de protection des personnes (CPP) (Ethique)
Comité en charge de l’examen de l’acceptabilité éthique des projets de recherche.
Les membres de CPP sont bénévoles, tenus au secret professionnel, indépendants vis à vis des investigateurs et des promoteurs et désignés par l’autorité administrative compétente. Le CPP veille à la protection des personnes qui se prêtent à une recherche médicale et veille au respect de la législation dans le cadre de la recherche médicale. L’avis favorable d’un CPP est obligatoire avant le début d’une recherche portant sur la personne humaine. Cet avis est demandé par le promoteur de la recherche auprès d’un CPP. Un seul avis est demandé pour chaque recherche. En cas de modification substantielle en cours de la recherche, une demande d’avis sur cette modification doit être envoyée au CPP.
-> Ethique
Comité indépendant de surveillance (Méthodologie)
Comité restreint constitué d'experts indépendants du promoteur habilité à proposer l'interruption d'un essai clinique.
Le comité indépendant de surveillance peut comporter des méthodologistes, statisticiens et cliniciens. Il peut être notamment interpellé en cas de survenue d'effets indésirables à une fréquence inattendue, et peut alors demander une levée d'aveugle, limitée au comité, pour comparer les différents groupes de traitement. Il rend alors son avis au promoteur qui décide de poursuivre ou non l'étude. L'existence d'un comité de surveillance doit être mentionné lors de la soumission du protocole au Comité de protection des personnes. Termes anglais: Data Safety Monitoring Board (DSMB) ou Data Monitoring Committee (DMC).
-> Effets indésirables
Commission de Transparence (Règlementation)
Commission chargée en France d’évaluer le service médical rendu (voir service médical rendu) d’un nouveau médicament, dont le fabricant demande un remboursement par la Sécurité sociale.
Le rôle de la commission de la Transparence est de comparer l’efficacité et la tolérance de médicaments d’une même classe thérapeutique (ce que ne fait pas la commission d’AMM).
-> Spécialité pharmaceutique, Autorisation de Mise sur le Marché
Commission Nationale Informatique et Liberté (CNIL) (Règlementation)
Commission veillant à la protection de la vie privée et des libertés des personnes notamment au regard du traitement des données dans le cadre des recherches.
-> Confidentialité, Respect de la personne, Base de données

Comparateur (Méthodologie)
Placebo ou traitement de référence utilisé dans un essai comparatif. Il est donné à un groupe (ou bras) alors que l’autre groupe (ou bras) reçoit le produit expérimental.
Comparateur peut être le traitement habituellement utilisé pour traiter la même maladie au même stade, le traitement de référence, s’il existe. En l’absence de traitement de référence, le produit expérimental peut être comparé à un placebo. Le recours à un placebo alors qu’existe un traitement de référence constitue une perte de chances éthiquement condamnable.
-> Placebo, Traitement de référence
Compliance (Méthodologie)
voir : Observance
Concomitant (Méthodologie)
Caractérise un état existant ou un évènement se produisant en même temps qu'un autre état ou évènement.
Le fait que deux éléments soient concomitants ne préjuge pas de l’existence d’un lien de causalité entre eux.
-> Causalité
Confidentialité (Ethique)
Ensemble des précautions pour qu'une information ne soit accessible qu'à ceux qui ont droit d'y accéder.
La règle du secret professionnel s’applique à toute personne qui participe de près ou de loin (médecin investigateur et son équipe, ARC, auditeurs, biométriciens...) à une recherche portant sur la personne humaine. Le secret professionnel s’applique même entre professionnels dès lors que le partage d’une information privée ne concourt pas à la santé de l’individu concerné. La confidentialité est un des aspects du « Respect de la personne ». Terme anglais : privacy
-> Ethique, Respect de la personne
Consentement éclairé (Ethique)
Acceptation libre et formellement exprimée d’une personne en vue de participer à une recherche dont elle a été informée des objectifs et contraintes.
Cette acceptation formelle ne doit être demandée qu’après avoir informé la personne en lui précisant les objectifs, les bénéfices, les risques et les inconvénients potentiels liés à l’essai ; la personne doit également être informée de ses droits et responsabilités, conformément à la version en vigueur de la déclaration d’Helsinki. Le consentement à participer à une recherche clinique peut être à tout moment retiré sans aucun préjudice pour la personne concernée.
-> Autonomie, Notice d’information
Contract Research Organisation (Règlementation)
Terme anglais voir : Organisme prestataire de services.
Contre-indication (Méthodologie)
Condition qui interdit une intervention médicale ou la prise d'un médicament.
Une contre-indication fait que l'état de santé du malade empêche un acte médical quel qu'il soit (administration d'un remède, intervention chirurgicale ou examen médical).
-> Indication, Résumé des caractéristiques du produit, Autorisation de Mise sur le Marché
Contrôle de la qualité (Règlementation)
Elément du système qui garantit des normes très strictes pendant la recherche, les essais et la production de médicaments.
Chaque étape du développement et de la production des médicaments est gérée par un système de Gestion de la Qualité. Les normes requises sont appelées système d'Assurance Qualité, tandis que le est la méthode utilisée pour garantir que les normes sont respectées à chaque étape. La gestion de la qualité pour la recherche clinique est connue sous le nom de Bonnes Pratiques Cliniques (BPC).
-> Bonnes pratiques cliniques
Convention d’Oviedo (Ethique)
Convention établie à l'échelle Européenne pour la protection des Droits de l’Homme et de la dignité de l’être humain à l’égard des applications de la biologie et de la médecine.
Cette convention est le seul instrument juridique contraignant international pour la protection des droits de l’Homme dans le domaine biomédical. Elle reprend les principes développés par la Convention européenne des Droits de l’Homme dans le domaine de la biologie et de la médecine.
-> Ethique
Coordonnateur (Méthodologie)
L’investigateur qui coordonne une recherche portant sur la personne humaine réalisée simultanément par plusieurs investigateurs sur plusieurs sites.
(étude multicentrique)
-> Essai multicentrique, Investigateur principal
Critère d’évaluation composite (Méthodologie)
Critère d’évaluation reposant sur un ensemble d’informations ou de mesures biologiques.
Les études portant sur des malades traités en unité de soins intensifs utilisent fréquemment des scores composites comme critère d’évaluation: score APACHE, score de Glasgow).
Critère d’évaluation de substitution (Méthodologie)
Critère qui ne mesure pas le résultat attendu du traitement mais un marqueur biologique dont on a montré qu’il lui était étroitement associé.
Par exemple, dans une étude portant sur l’augmentation de la survie de malades sur un temps long, l’évaluation de l’efficacité sur ce critère imposerait une durée d’étude considérable. La mesure d’un paramètre biologique ayant une bonne valeur pronostique sur l’issue individuelle de la maladie permet alors d’évaluer plus précocement l’effet du traitement. Le rapport entre le nombre de deux types de cellules sanguines (lymphocytes T4 et T8) a été largement utilisé comme critère de substitution à la survie dans l’évaluation de traitements du SIDA.
-> Biomarqueur
Critère d’évaluation principal (Méthodologie)
Critère d’évaluation permettant de répondre à l’objet principal de l’étude.
C’est en fonction, d’une part de la différence attendue sur ce critère entre le groupe traité par le produit expérimental et son comparateur, d’autre part de la variabilité de ce critère dans la population étudiée, qu’est calculé le nombre de personne requis pour une recherche donnée.
-> Effectif, Objet de l’étude, Puissance
Critère de jugement (Méthodologie)
voir : Critère d’évaluation
Critères d’évaluation (Méthodologie)
Critères permettant de répondre à l’objet de l’étude.
Ces critères sont définis lors de la planification de la recherche et sont décrits dans le protocole. Les qualités attendues d’un critère d’évaluation sont la spécificité, la sensibilité, et la reproductibilité. Le choix des critères d’évaluation conditionne celui des examens pratiqués pour les obtenir et donc les contraintes liées à la recherche. Synonyme : critère de jugement.
-> Critères d’évaluation secondaires, Objectif principal, Effectif
Critères d’évaluation secondaires (Méthodologie)
Critères permettant de documenter les objectifs secondaires fixés dans l’étude.
Les critères secondaires permettent de documenter les objectifs secondaires fixés dans l’étude. Les critères d’évaluation portant sur la tolérance du produit et/ou qualité de vie font le plus souvent partie des critères d’évaluation secondaires.
-> Qualité de vie
Critères d’inclusion (Méthodologie )
Critères définissant les caractéristiques des sujets ou des patients qui peuvent participer à une étude.
Les critères d’inclusion peuvent concerner des caractéristiques individuelles (âge, sexe, …), la maladie affectant la personne (stade, type génétique…), voire des exigences règlementaires (affiliation à un régime de sécurité sociale…).
-> Eligibilité, Admissibilité, Critères de non-inclusion
Critères de non inclusion (Méthodologie)
Critères définissant les personnes non éligibles pour une recherche clinique.
(Gravidité, traitement habituel par un autre médicament, traitement antérieur…). Ils servent notamment à protéger le participant à l’étude (éviter les contre-indications).
-> Éligibilité, Admissibilité, Critères d’inclusion
Cross over (Méthodologie)
Terme anglais, voir : Essai croisé
Début d'un essai clinique (Règlementation)
Signature du consentement éclairé par le premier participant, sauf si le protocole donne une autre définition.
Déclaration d’Helsinki (Ethique)
Enoncé de principes éthiques applicables à la recherche médicale impliquant des êtres humains, y compris la recherche sur du matériel biologique humain et sur des données identifiables.
Ce texte émane de l’Association Médicale Mondiale, sa première version date de 1964 et il est régulièrement révisé (9ème version en 2013) https://www.wma.net/fr/policies-post/declaration-dhelsinki-de-lamm-principes-ethiques-applicables-a-la-recherche-medicale-impliquant-des-etres-humains
-> Ethique
Déclaration universelle des droits de l’homme (Règlementation)
Document international qui définit les droits de base et les libertés fondamentales revenant à tous les êtres humains.
Elle a été adoptée par l’Assemblée générale des Nations Unies en 1948, à la suite de l’expérience et des atrocités de la Seconde Guerre mondiale. Elle représentait la première expression mondiale des droits revenant à tous les êtres humains, sans distinction de nationalité, de lieu de résidence, de genre, d’origine nationale ou ethnique, de couleur, de religion, de langue ou de tout autre statut. La Déclaration a été inscrite dans la législation sous plusieurs formes et a inspiré plus de 80 déclarations et traités internationaux des droits de l’homme qui, ensemble, constituent un système complet et juridiquement contraignant afin de promouvoir et protéger les droits de l’homme. Le texte complet de la Déclaration est publié par les Nations Unies sur son site : http: //www.un.org/fr/universal-declaration-human-rights/index.html
-> Ethique
Délai de réflexion (Ethique)
Temps séparant la délivrance de l’information et la signature du consentement ou l’expression de la non-opposition.
Sa durée doit être suffisante pour permettre un choix éclairé. Néanmoins, ce délai peut être réduit au seul temps de l’information préalable lorsque le traitement à l’étude ne peut être médicalement différé.
-> Autonomie
Dénomination Commune Internationale (DCI) (Pharmacologie)
Dénomination d'un médicament attribuée par l'Organisation Mondiale de la Santé (OMS) en collaboration avec les commissions nationales de nomenclature.
La DCI identique dans le monde entier (à quelques nuances liées aux règles de prononciation) se distingue des noms commerciaux (noms de fantaisie) qui peuvent varier d’un pays à l’autre, ou d’un fabricant à un autre dans un même pays. Les DCI sont formées en utilisant des racines communes dans une même famille de médicament favorisant ainsi leur identification, par exemple propanolol, aténolol, oprénolol sont trois bêtabloquants utilisés dans le traitement de l'hypertension artérielle.
-> Spécialité pharmaceutique, Médicament
Déontologie (Ethique)
L’ensemble des principes, règles et usages que doit respecter un professionnel dans l’exercice son activité.
-> Ethique
Désignation médicament orphelin (Règlementation)
Statut administratif attribué par l’EMA à une substance en cours d’étude dont l’utilisation future comme traitement d’une maladie rare parait «plausible ».
Il s’agit de médicaments en cours de développement, destinés à traiter, à prévenir, ou à diagnostiquer les maladies rares (prévalence< 1/2 000) pour lesquelles il n’existe aucune méthode satisfaisante de diagnostic, de prévention ou de traitement autorisé en Europe pour la maladie considérée ; ou s’il en existe, le médicament orphelin procurera un bénéfice notable aux personnes malades par rapport aux méthodes déjà existantes. La « désignation » permet de bénéficier des mesures incitatives au laboratoire développant ce médicament ; lors de son développement, l’assistance au protocole par l’EMA pour guider le demandeur dans ses choix, l’accès à la procédure centralisée de demande d’AMM, et des réductions de charge ; en cas d’obtention d’une AMM, une exclusivité commerciale de 10 ans au sein de l’Union Européenne après obtention de cette AMM.
-> Médicament orphelin, European Medicine Agency, Exclusivité commerciale
Dispensation des médicaments (Règlementation)
Délivrance d’un médicament à titre clinique ou dans une recherche clinique.
A l’hôpital cette responsabilité incombe au pharmacien hospitalier.
Dispositif médical (DM) (Règlementation)
Instrument, appareil, équipement ou encore un logiciel destiné, chez l’homme, au diagnostic, à la prévention, au contrôle, au traitement d’une maladie...
Distribution (Pharmacologie)
Transport du médicament au niveau sanguin (phase plasmatique) puis sa diffusion dans les tissus (phase tissulaire).
-> Pharmacocinétique, Absorption, Distribution, Métabolisme, Elimination
Dose maximale tolérée (Phamacologie)
Dose la plus élevée d'un médicament ou d'un traitement donné/administré sans entraîner des effets secondaires inacceptables.
Elle est déterminée lors d'essais cliniques en testant des doses croissantes sur différents groupes d'individus jusqu'à ce que la plus haute dose avec des effets secondaires tolérés soit trouvée.
-> Toxicité, Effet indésirable
Double aveugle (Méthodologie)
voir : Double insu
Double insu (Méthodologie)
Procédure expérimentale où ni le participant ni l'équipe de recherche ne connaissent le groupe de traitement auquel appartient chaque participant.
L'insu permet d'éviter un biais qui peut être intentionnel ou non si l'équipe de recherche ou les participants savent à quel groupe ces derniers appartiennent (biais d'évaluation).
-> Insu, Simple insu, Essai en ouvert
Effectif (Méthodologie)
Nombre total de personnes participant à une recherche.
L’effectif d’une recherche ou de chaque groupe est généralement représenté par la lettre « n ».
-> Puissance, Valeur de p
Effet indésirable (Phamacologie)
Toute réaction nocive, survenant chez une personne traitée par un médicament et attribuable à ce dernier.
C’est ce lien causal qui différencie un évènement indésirable (concomitant de la prise du médicament sans lien causal établi) d’un effet indésirable (lien causal établi).
-> Evènement indésirable, Toxicité
Effet indésirable grave (Phamacologie)
Effet indésirable qui nécessite l’hospitalisation, met la vie en danger, provoque un handicap ou une malformation congénitale.
Il doit impérativement être immédiatement reporté à l’ANSM.
-> Evènement indésirable, Toxicité
Effet nocebo (Pharmacologie)
Effet indésirable lié à l’administration d’une substance, indépendamment ou en plus de ses propriétés pharmacologiques.
L’effet nocebo peut donc s’observer avec l’administration d’une substance dénuée de tout effet pharmacologique comme un placebo.
-> Effet placebo, Comparateur
Effet placebo (Phamacologie)
Effet thérapeutique lié à l’administration d’une substance, indépendamment ou en plus de ses propriétés pharmacologiques.
L’effet placebo peut donc s’observer avec l’administration d’une substance dénuée de tout effet pharmacologique. Cet effet est influencé par la confiance qu’a le patient dans les effets potentiels d’un médicament.
-> Placebo, Effet nocebo, Comparateur
Effet secondaire (Phamacologie)
Effet provoqué par un médicament différent de l'effet recherché.
Pas nécessairement nocifs, les effets secondaires sont généralement connus pour des molécules qui sont sur le marché, apparaissent dans le résumé des caractéristiques du produit et sont notifiés sur la notice du médicament.
-> Notice, Résumé des caractéristiques du produit
Efficacité (Phamacologie)
Effets bénéfiques obtenus avec un médicament.
Dans un essai clinique, l’efficacité du produit expérimental est comparée à un autre traitement ou à placebo.
-> Essai comparatif, Rapport bénéfice/risque
Éligibilité (Méthodologie)
Est « éligible » à une recherche une personne qui répond à tous les critères d’inclusion et à aucun critère de non inclusion de cette recherche.
Les deux conditions pour participer à une recherche sont l’éligibilité et l’expression de son consentement à participer.
-> Critères d’inclusion, Admissibilité
Elimination (Pharmacologie)
Dernière phase du devenir du médicament dans l’organisme.
Cette élimination peut concerner le médicament sous forme inchangée, ou nécessiter qu’il ait été préalablement métabolisé par le foie.
-> Pharmacocinétique, Absorption, Distribution, Métabolisme, Elimination
Essai clinique (Règlementation)
Recherche portant sur la personne humaine, concernant un produit sans autorisation de mise sur le marché, ou nécessitant des examens supplémentaires.
Il est défini par la directive européenne de 2014 comme : une situation expérimentale réalisée sur l’être humain visant à déterminer ou à confirmer les effets cliniques, pharmacologiques, pharmacodynamiques ou à mettre en évidence tout effet indésirable, ou à étudier l’absorption, la distribution le métabolisme et l’élimination de produits dans le but de s’assurer de leur innocuité ou de leur efficacité.
-> Recherche clinique
Essai comparatif (Méthodologie)
Essai clinique qui compare au moins deux traitements, celui que l’on évalue et un comparateur.
Le comparateur peut être un traitement de référence, reconnu comme efficace et utilisé habituellement pour soigner la même maladie, ou un placebo. Terme anglais : Controlled trial.
-> Comparateur, Placebo
Essai croisé (Méthodologie)
Essai clinique qui compare l’effet de x médicaments pris à des périodes différentes chez un même patient dans un ordre défini lors de la randomisation.
Lors d’une première période de traitement, le patient prendra le traitement A. Puis après une période sans traitement (intervalle libre ou wash out en anglais), le même patient prendra le traitement B. L’intérêt de ce type d’étude réside dans le fait que chez un même patient est mesuré l’effet de A et B (le patient est son propre témoin). Terme anglais : cross-over.
-> Essai comparatif, Essai en groupes parallèles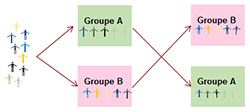
Essai en groupes parallèles (Méthodologie)
Essai clinique au cours duquel sont formés deux ou plusieurs groupes de sujets ou de patients qui diffèrent par leur traitement.
Dans l’exemple de deux traitements A et B, le groupe 1 reçoit le traitement A et le groupe 2 reçoit le traitement B. Cette attribution se fait le plus souvent par tirage au sort (principe de la randomisation). Les deux groupes sont suivis de la même façon (examens, durée, nombre de visites).
-> Essai croisé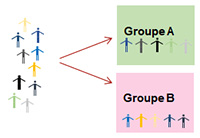
Essai en ouvert (Méthodologie)
Essai clinique dans lequel l'investigateur et/ou la personne qui y participe connaissent le traitement qui est donné à cette personne lors de l’étude.
Inverse : essai en insu.
-> Insu
Essai monocentrique (Méthodologie)
Essai qui est réalisé dans un seul centre.
-> Essai multicentrique
Essai multicentrique (Méthodologie)
Essai réalisé selon le même protocole dans plusieurs lieux géographiques.
La recherche est placée sous la responsabilité d’un investigateur coordonnateur. Dans chaque centre est désigné un investigateur principal qui tient à jour, pour son centre, la liste des investigateurs et personnes associés à la recherche.
-> Essai monocentrique, Investigateur coordinateur
Essai randomisé (Méthodologie)
Essai dont les traitements sont attribués par tirage au sort (randomisation).
La randomisation et l’insu sont deux procédures limitant les risques de biais des recherches cliniques.
-> Randomisation, Insu, Biais
Ethique (Ethique)
Science de la morale et des mœurs.
C'est une discipline philosophique qui réfléchit sur les finalités, sur les valeurs de l'existence, sur les conditions d'une vie heureuse, sur la notion de "bien" ou sur des questions de mœurs ou de morale.
-> Bienveillance, Autonomie, Justice distributive, Déclaration d’Helsinki
European Medicine Agency (EMA) (Règlementation)
Agence Européenne du Médicament. Sa principale mission est la protection et la promotion de la santé publique et animale à travers l’évaluation et la supervision des médicaments à usage humain et vétérinaire.
Parallèlement aux agences nationales du médicament, l’EMA peut délivrer des Autorisations de Mise sur le Marché. L’EMA (au travers de commissions spécialisées) a la responsabilité des autorisations concernant certaines catégories de médicaments, comme les médicaments orphelins (COMP), les médicaments pédiatriques (PDCO), et les biothérapies dites innovantes (CAT).
-> AMM centralisée, Médicaments orphelins, Médicaments pédiatriques
Événement indésirable (Pharmacologie)
Toute manifestation nocive chez un participant auquel un médicament est administré, et qui n'est pas nécessairement liée à ce traitement.
Événement indésirable grave : événement entrainant le décès, l’hospitalisation ou une prolongation d’hospitalisation, une incapacité ou un handicap important ou durable, une mise en danger la vie du sujet, une anomalie ou malformation congénitale.
Événement indésirable non grave : tout autre événement indésirable.
Dans un essai clinique, les évènements indésirables graves doivent être immédiatement signalés au promoteur. Le promoteur a l’obligation de déclarer à l’ANSM, dans un délai court (7 jours en cas de décès), toutes les suspicions d’effets indésirables graves inattendus pour les recherches portant sur les médicaments.
Examen médical préalable (Règlementation)
Examen médical auquel est soumis un participant potentiel afin de vérifier que son état de santé est en accord avec les critères d’inclusion et de non inclusion.
Cet examen est obligatoire (article L1211-11 du Code de la Santé publique). Ses résultats doivent être communiqués aux participants soit directement, soit s’ils le souhaitent, par l’intermédiaire du médecin de leur choix.
-> Eligibilité, Inclusion
Exclusivité commerciale (Règlementation)
Dispositif administratif interdisant pendant une durée spécifiée, la mise sur le marché d’un nouveau médicament, équivalent, pour les mêmes indications que celui bénéficiant de cette exclusivité.
L’exclusivité commerciale fait partie des mesures incitatives Européennes proposées pour la mise au point de médicaments orphelins et de médicaments pédiatriques. Cette mesure ne s’applique pas dans le cas où le nouveau médicament présente des avantages substantiels, en termes d’efficacité ou de moindre toxicité, par rapport au médicament ainsi protégé. L’exclusivité commerciale se distingue de la protection industrielle en ce qu’elle débute à la date de l’AMM et non de celle du dépôt de brevet.
-> Désignation, Médicament orphelin, Médicament pédiatrique
Expérimentation animale (Méthodologie)
Utilisation d'animaux, pour mieux comprendre la physiologie d'un organisme et ses réponses à divers facteurs ou substances, pour en tester notamment l'innocuité ou la toxicité pour tenter de prévoir ce qui se passe chez l'Homme.

Fichier national des personnes qui se prêtent à des recherches impliquant la personne humaine (Règlementation)
Fichier national qui recense les personnes qui ne présentent aucune affection et se prêtent volontairement à ces recherches ainsi que les personnes malades lorsque l'objet de la recherche est sans rapport avec leur état pathologique.
Le comité de protection des personnes peut décider dans certains cas, compte tenu des risques et des contraintes que comporte la recherche impliquant la personne humaine, que les personnes qui y participent doivent être également inscrites dans ce fichier.(Code de Santé Publique, article L1121-16 du Code de Santé Publique).
-> Comité de protection des personnes
Fin d'un essai clinique (Méthodologie)
Dernière visite du dernier participant, ou un moment ultérieur défini par le protocole.
Fraude (Méthodologie)
Acte intentionnel de duperie dans la production, l’analyse ou la présentation des résultats d’une recherche.
Une fraude dans la production des résultats peut consister en l’invention, la modification ou la suppression de résultats. Dans l’analyse, l’utilisation délibérée de méthodes inadaptées, l’analyse sur des sous-groupes choisis après connaissance des résultats, enfin dans la présentation des résultats par des conclusions injustifiées par les résultats. La fraude n'inclut pas des erreurs naïves ou relevant d’incompétence, ou des processus médiocres de recherche, sauf si ceci a été fait avec une intention de tromper. Dans le domaine de la recherche, la fraude peut avoir un impact financier sur le promoteur, avoir de lourdes conséquences pour la crédibilité d'une étude, voire même mener des patients à accéder à des traitements inefficaces ou nocifs.
Gène (Biologie)
Unité d’information génétique qui contient l'information nécessaire à la fabrication d’une protéine ou d'une autre molécule (comme l'ARN), qui sont essentielles à la croissance et au fonctionnement d'un organisme.
Générique (Règlementation)
Qualifie une spécialité pharmaceutique en reproduisant une autre déjà autorisée, et ayant les mêmes caractéristiques.
-> Résumé des caractéristiques du produit, Autorisation de Mise sur le Marché
Génie génétique (Phamacologie)
Ensemble de techniques biologiques permettant de faire produire des molécules biologiques, notamment à visée thérapeutique, par des organismes vivants, animaux ou microorganismes.
Le génie génétique est une alternative à la synthèse chimique, en particulier pour des molécules de très grande taille (hormones, enzymes, anticorps,…).
Génome (Biologie)
Ensemble des gènes d’un organisme ou ensemble de son ADN.
Génomique (Biologie)
Branche de la biologie qui étudie le génome entier d'un organisme par séquençage, assemblage et analyse de la fonction de son ADN.
Les avancées de la génomique ont permis de réaliser des progrès considérables dans la compréhension de diverses maladies. La recherche en génomique peut permettre de mettre au point des stratégies thérapeutiques plus efficaces et de meilleurs outils de prise de décision pour les patients et les professionnels de santé.
-> Technologies « omiques ».
Génotype (Biologie)
Ensemble des gènes codant pour des caractéristiques ou des aptitudes d’un individu.
-> Phénotype
Groupe de traitement (Méthodologie)
Groupe de participants à une recherche clinique recevant le même traitement.
Le traitement attribué à un groupe peut être le produit évalué ou un comparateur (traitement de référence ou placebo). Dans un essai comportant plus de deux groupes, les différents traitements peuvent être différents dosages d’un même produit ou différents produits. Dans une étude randomisée, l’appartenance de chaque sujet à un groupe donné est déterminée par tirage au sort. Synonyme : Bras.
-> Essai comparatif, Comparateur, Randomisation
Haute Autorité de Santé (Règlementation)
Autorité publique indépendante française qui contribue à la régulation du système de santé.
Elle exerce ses missions dans les champs de l'évaluation des produits de santé, des pratiques professionnelles, de l’organisation des soins et de la santé publique.
Hétérogénéité (Statistique)
La diversité clinique (ou hétérogénéité) est la variabilité entre les patients ou les interventions étudiées, ou entre les résultats que les études mesurent.
Lorsque différentes études sont comparées, il est important de garder à l'esprit qu'il existe plusieurs types d'hétérogénéité. La diversité méthodologique (ou hétérogénéité) fait référence à la variation de la méthodologie des études et du risque de biais entre les études. La diversité clinique et/ou méthodologique peut conduire à des différences d'application des statistiques aux différentes études (hétérogénéité statistique). Les progrès réalisés en sciences médicales améliorent notre compréhension de l'hétérogénéité parmi les patients atteints de la même maladie. Les différences de réponses des patients au traitement et le risque de réactions indésirables sont étudiés au niveau moléculaire, et des traitements ciblés sont, par conséquent, développés pour les différents sous-groupes de patients.

Imputabilité (Pharmacologie)
Etablissement d’un lien de causalité entre une intervention et un évènement observé.
L’analyse d’imputabilité repose sur des critères chronologiques, la mise en évidence de mécanismes biologiques pour distinguer un lien de causalité entre intervention et évènement d’une simple concomitance. A la suite de la prise d’un médicament, l’imputabilité est requise pour qualifier un évènement indésirable d’effet indésirable.
-> Evènement indésirable, Effet indésirable, Concomitance
In silico (Méthodologie)
Néologisme formé sur le modèle de « in vivo » et « in vitro », désignant des études réalisées par simulation informatique.
-> In vitro, In vivo
In vitro (Méthodologie)
En latin « dans le verre », qualifie des expérimentations réalisées sans recourir à des êtres vivants.
-> In vivo, In silico, Phase préclinique
In vivo (Méthodologie)
En latin « chez le vivant », qualifie des expérimentations recourant à des êtres vivants.
-> In vitro, In silico, Phase préclinique
Inclusion (Méthodologie)
Processus établissant la participation d’une personne à une recherche clinique.
L’inclusion d’un participant repose à la fois sur son éligibilité et son consentement éclairé.
-> Eligibilité, Consentement éclairé, Participant
Indemnisation (Règlementation)
Compensation financière accordée à des participants à une recherche clinique.
La loi française pose en principe général qu’il n’y a pas de contrepartie financière pour les participants à une recherche portant sur la personne humaine, hormis le remboursement des frais exposés liés à la recherche (exemple : frais de transport), (article L1121-11 du Code de Santé Publique).
Toutefois, il est possible de verser aux participants à la recherche une indemnité de compensation des contraintes subies selon certaines conditions :
l’indemnité est versée par le promoteur,
Le montant annuel de l’indemnité est plafonné par arrêté ministériel.
L’indemnité est interdite pour :
les mineurs,
les majeurs hors d’état d’exprimer leur consentement,
les prisonniers,
les personnes avec mesure de protection légale ou hospitalisées sans leur consentement,
les personnes admises dans un Établissement sanitaire et social à d’autres fins que la recherche.
Le montant de l’indemnité prévue et réellement versée doit être communiqué au CPP ainsi que les modalités d’indemnisation des sujets ou des patients. L’indemnité ne constitue ni un salaire ni des honoraires. Elle n’est donc pas soumise à imposition ni au versement de charges sociales par le promoteur ou la personne participant à une recherche portant sur la personne humaine.
Indication (Médecine)
Situation pathologique justifiant l'utilisation d'un traitement.
L'autorisation de mise sur le marché définit précisément les indications et les posologies d'un médicament. Lorsqu'un médicament est utilisé en dehors des indications précisées dans l'AMM, on parle d'utilisation "hors AMM". L’indication peut correspondre à une maladie, ou à une situation plus restrictive, stade de la maladie, type génétique, etc.
-> Autorisation de Mise sur le Marché, Contre-indication, Résumé des caractéristiques du produit
Information des participants (Méthodologie)
Elément capital de la protection de ces personnes participant à une recherche, indispensable avant le recueil d'un consentement.
L'information (Code de Santé Publique L1222-1) porte notamment sur :
1° L'objectif, la méthodologie et la durée de la recherche,
2° Les bénéfices attendus, les contraintes et les risques prévisibles, y compris en cas d'arrêt de la recherche avant son terme,
3les éventuelles alternatives médicales,
4° les modalités de prise en charge médicale prévues en fin de recherche, si une telle prise en charge est nécessaire, en cas d'arrêt prématuré de la recherche, et en cas d'exclusion de la recherche,
5° L'avis du CPP let l'autorisation de l'autorité compétente,
6° Le cas échéant, l'interdiction de participer simultanément à une autre recherche,
6° bis Pour les recherches à finalité commerciale, les modalités de versement de,
7° Le cas échéant, la nécessité d'un traitement des données personnelles L’information doit être donnée par le médecin investigateur ou un médecin le représentant et elle doit être objective, loyale et compréhensible par le sujet.
Inocuité (Pharmacologie)
Absence de toxicité.
-> Toxicité
Inspection (Règlementation)
Activité menée par une autorité compétente, l’ANSM pour les essais cliniques, consistant à procéder à l'examen officiel des documents, installations, enregistrements, systèmes d'assurance qualité et de tout autre élément qui, de l'avis de l'autorité compétente, ont trait à l'essai clinique.
L'inspection peut concerner le site d'essai clinique, les locaux du promoteur et/ou de l'organisme de recherche sous-traitant ou tout autre établissement que l'autorité compétente juge nécessaire d'inspecter.
-> Bonnes pratiques cliniques, Fraude, Site d’essai clinique, Agence Nationale de Sécurité du Médicament et des Produits de Santé
Inserm (Règlementation)
Organisme public français de recherche dédié à la santé humaine.
Créé en 1964, l'Inserm assure la coordination stratégique, scientifique et opérationnelle de la recherche biomédicale, l'expertise et la veille scientifiques.
Institutional Review Board (IRB) (Ethique)
voir : Comité d'éthique de la recherche
Insu (Méthodologie)
Façon de garantir que les personnes impliquées dans une étude de recherche, comme les participants à des essais cliniques, ne savent pas à quel groupe de l'essai ils appartiennent.
Cette méconnaissance prévue et organisée par le protocole d’une étude, de la réalité ou de la nature du traitement auquel est exposé un sujet ou un groupe a pour but d’éviter que cette connaissance n’influe, directement ou indirectement sur l’évaluation des traitements étudiés. Dans un essai comportant, par exemple, un groupe de traitement et un groupe placebo, l'insu signifie que les participants ne savent pas s'ils reçoivent le traitement ou le placebo. Le terme « simple insu » s'emploie parfois pour décrire les études dans lesquelles les participants ne connaissent pas le groupe auquel ils appartiennent, mais que l’équipe de recherche le sait. La mise en œuvre de l’insu exige la préparation de formes pharmaceutiques spécifiques à l’essai, parfaitement identiques dans leur présentation, dont la seule différence (uniquement identifiable par un code) est la nature des ingrédients, actifs ou non que l’on veut comparer. Le code, connu seulement de l’organisateur de l’essai est levé en fin de période d’étude ou avant, si des impératifs de sécurité l’exigent. Avant le début de l’essai, au moment de l’information et de la signature du consentement, le malade a connaissance des différentes possibilités de traitement qui peuvent lui être appliquées. Un essai en insu est l'inverse d'un essai dit « ouvert » ou « en ouvert ». Synonyme : étude en aveugle.
-> Double insu, Simple insu, Essai en ouvert, Placebo
International Conference on Harmonisation (ICH) (Méthodologie)
Structure internationale qui rassemble les autorités de réglementation et les représentants de l'industrie pharmaceutique d'Europe, du Japon et des États-Unis pour discuter des aspects scientifiques et techniques de l'enregistrement des médicaments.
La mission de la ICH est de parvenir à l'harmonisation des données et des règlements et de s'assurer ainsi de la sûreté, de la qualité et de l'efficacité des médicaments développés et enregistrés par les différents pays participants Elle produit des recommandations de bonnes pratiques, concernant, la qualité, la sécurité, et l'efficacité de l'évaluation des médicaments.
-> Bonnes pratiques cliniques
Interruption prématurée de traitement (Méthodologie)
Arrêt de la prise du traitement par un participant à une recherche clinique avant la fin prévue de l’essai, quelle qu’en soit la cause.
Cette interruption n’empêche pas la poursuite de la participation à la recherche à condition que le sujet ne retire pas son consentement, ce sujet sera pris en compte dans une analyse dite « en intention de traiter », pas dans une analyse dite « per protocole". Les participants ayant interrompu leur traitement sont à distinguer des participants « perdus de vue».
-> Analyse en intention de traiter, Analyse per protocole, Perdu de vue
Intervalle de confiance (Statistique)
Plage de valeurs calculée dans laquelle une valeur individuelle à une probabilité connue de se trouver.
Dire que l’intervalle de confiance à 95% de la réduction de la pression artérielle après un traitement donné est entre 5,4 et 9,4 mm Hg (noté IC95 = 5,4 - 9,4 mm Hg) signifie qu’un individu de la population étudiée à 95% de chance pour que la diminution de sa propre pression artérielle soit comprise entre ces deux valeurs. Cette grandeur informe à la fois sur la moyenne de l’effet et son hétérogénéité.
-> Moyenne, Hétérogénéité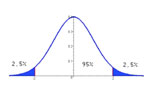
Intervalle libre (Méthodologie)
Période pendant laquelle le sujet ou le patient ne reçoit aucun traitement. Il s’agit d’une période intercalée entre deux périodes de traitements.
Terme anglais, période de wash-out.
-> Essai croisé
Investigateur (Règlementation)
Médecin qui dirige et surveille la réalisation de la recherche portant sur la personne humaine (article L1121-3 du Code de la Santé publique).
Ce médecin doit justifier d’une expérience appropriée. En fonction des domaines de recherche, l’investigateur peut également être une sage-femme ou un dentiste.
-> Investigateur principal, Investigateur coordinateur
Investigateur coordonnateur (Règlementation)
Investigateur qui assure la coordination de l’ensemble des centres avec le promoteur, le CPP, etc., dans le cadre d'une recherche multicentrique.
-> Essai multicentrique
Investigateur principal (Règlementation)
Investigateur responsable d'une équipe d'investigateurs chargée de la conduite d'un essai clinique sur un site d'essai clinique.
Les autres médecins qui participent à la recherche sont appelés investigateurs associés ou collaborateurs.
Justice distributive (Ethique)
Théorie philosophique traitant de la distribution des avantages répartis entre les membres d'une communauté.
Mentionnée initialement par Aristote, elle a été développée au XXème siècle par J. Rawls et fait partie avec la bienfaisance et le respect de la personne des fondements éthiques de la recherche décrits dans ses textes fondateurs (Rapport Belmont, Déclaration d’Helsinki, Convention d’Oviedo).
-> Ethique, Rapport Belmont, Déclaration d’Helsinki, Convention d’Oviedo
Lieu de recherche (Règlementation)
Lieu disposant des moyens humains, matériels et techniques adaptés à la recherche et compatibles avec les impératifs de sécurité des personnes qui s'y prêtent.
Ce lieu doit être autorisé, à cet effet, pour une durée déterminée, lorsqu'il s'agit de recherches réalisées en dehors des lieux de soins. Cette autorisation est accordée par le directeur général de l'agence régionale de santé ou par le ministre de la défense, si le lieu relève de son autorité. La première administration d'un médicament à l'homme dans le cadre d'une recherche ne peut être effectuée que dans des lieux ayant obtenu l'autorisation mentionnée au deuxième alinéa du présent article (article L1121-13 du Code de Santé Publique).
-> Investigateur principal
Maladie rare (Médecine)
Dans les pays de l'Union européenne (UE), toute maladie touchant moins de 5 personnes sur 10 000.
Ce sont des maladies le plus souvent génétiques qui mettent la vie en danger ou entraînent une invalidité chronique. On estime qu'aujourd'hui, dans l'UE, 5 000 à 8 000 maladies rares touchent 6 à 8 % de la population, soit entre 27 et 36 millions de personnes.
Médecine personnalisée (Médecine)
Modèle médical qui se propose d'adapter les décisions et pratiques médicales ainsi que les traitements à chaque patient.
Elle fait appel à des médicaments ciblés visant des molécules précises impliquées dans la maladie du patient et tient compte des informations génétiques, cliniques, environnementales ainsi que sur le mode de vie du patient. Le but de ce type de médecine est de sélectionner les traitements les plus adaptés à chaque patient, afin de garantir les meilleurs résultats et de réduire le risque d'effets secondaires. Les progrès réalisés dans la compréhension du lien entre la génomique (et d'autres facteurs moléculaires) et la maladie est une part importante du développement de la médecine personnalisée. Aussi, les laboratoires pharmaceutiques produisent-ils déjà certains médicaments ciblés. Synonyme : Médecine de précision.
-> Sous groupe, Effet secondaire, Médecine stratifiée.
Médecine stratifiée (Médecine)
Médecine basée sur l'identification de sous-groupes de patients dont les mécanismes de la maladie, leur sensibilité à une maladie particulière ou leur réponse à un médicament sont différents.
L'objectif de la médecine stratifiée est d'offrir le traitement le plus susceptible d'apporter un bénéfice ou d'éviter une réaction indésirable. La médecine personnalisée approfondit cette approche en utilisant des médicaments ciblés et en tenant compte d'informations sur le patient telles que son génotype et son mode de vie au moment du choix du meilleur traitement.
-> Sous-groupe, Médecine personnalisée
Médicament (Pharmacologie)
Substance ou composition présentée comme ayant des propriétés curatives ou préventives à l'égard des maladies humaines, ainsi que tout produit pouvant être administré à l'homme en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier ses fonctions organiques.
Médicament biologique (Pharmacologie)
Un médicament biologique, ou biomédicament est un médicament produit par un organisme vivant, spontanément ou par génie génétique, par opposition aux substances produites par voie chimique, souvent qualifiées de petites molécules.
Grâce aux progrès du génie génétique, les biomédicaments occupent une part croissante de l’arsenal thérapeutique : hormones comme l’insuline, antigènes monoclonaux, enzymes de substitution, etc.
-> Génie génétique, Petite molécule
Médicament biosimilaire (Pharmacologie)
Médicament biologique similaire à un autre médicament biologique qui a déjà reçu l’autorisation de mise sur le marché.
Le médicament biologique existant se nomme « médicament de référence ». Les biosimilaires ne peuvent être mis sur le marché qu’après expiration du brevet du médicament de référence, mais ils peuvent néanmoins être développés avant ce délai. Un médicament biosimilaire et son médicament de référence doivent avoir le même profil d’innocuité et d’efficacité. Les médicaments biosimilaires sont développés de telle sorte qu’ils aient le même mécanisme d’action et traitent les mêmes maladies que le médicament de référence. La fabrication des médicaments biosimilaires est régie par les mêmes normes de Bonnes pratiques de fabrication (BPF) de l’UE que pour tout autre médicament biologique. Les médicaments biosimilaires peuvent représenter une alternative moins coûteuse que les médicaments biologiques existants qui ont perdu leurs droits d’exclusivité.
-> Biomédicament, Générique
Médicament expérimental (Pharmacologie)
Médicament expérimenté dans une recherche portant sur la personne humaine,
y compris les médicaments bénéficiant déjà d'une autorisation de mise sur le marché (AMM) mais utilisés et présentés ou conditionnés différemment de la spécialité autorisée, ou utilisés pour une indication non autorisée ou en vue d'obtenir de plus amples informations sur la forme de la spécialité autorisée.
Métabolisme (d’un médicament) (Pharmacologie)
Ensemble des transformations que subit un médicament introduit dans un organisme vivant.
L’organe le plus important qui réalise ces biotransformations chez l’homme est le foie mais de nombreux tissus possèdent les enzymes nécessaires au métabolisme des médicaments (ex : paroi intestinale, cellules du néphron).
-> Pharmacocinétique, Absorption, Distribution, Elimination
Modification substantielle (Règlementation)
Changement apporté à n'importe quel aspect de l'essai clinique susceptible d'avoir une incidence substantielle sur la sécurité ou les droits des participants ou sur la fiabilité et la robustesse des données obtenues lors de l'essai clinique.
-> Protocole
Moniteur d’essai clinique (Méthodologie)
voir : Assistant de recherche clinique.
Morbidité (Médecine)
Etat de maladie.
Par extension ce terme désigne aussi le nombre de personnes souffrant d'une maladie donnée pendant un temps donné, dans une population. En épidémiologie on préfère distinguer l'incidence (nouveaux cas observés pendant une période donnée ramenés à la population) et la prévalence (nombre de personnes malades à un moment donné, ramenés à la population). Le terme morbidité s'oppose généralement à mortalité.
-> Santé publique
Mortalité (Médecine)
Phénomène de la mort considéré comme évènement.
Le taux de mortalité représente le rapport entre le nombre de personnes décédées et l’ensemble de la population concernée, pendant un temps donné. Il peut être global ou ne concerner que les décès dus à une cause ou une maladie donnée.
-> Santé publique
NOAEL (Pharmacologie)
No Observable Adverse Effect Level.
Voir Seuil « aucun effet indésirable observé.»
-> Toxicité, Effet indésirable
Non-malfaisance (Ethique)
Attitude visant à éviter de nuire.
La non-malfaisance est au cœur de l'éthique médicale et fait partie du Serment d'Hippocrate (un serment prêté dans de nombreux pays par les médecins nouvellement diplômés). Un exemple d'action non-malfaisante consisterait à interrompre une médication connue pour être dangereuse ou à refuser d'administrer un médicament à un patient si son efficacité n'a pas été établie. Cependant, des dilemmes éthiques surviennent souvent. Dans de nombreuses situations médicales, la non-malfaisance doit être équilibrée avec le principe de bienfaisance (une action réalisée pour le bien des autres). Par exemple, de nombreux médicaments bénéfiques peuvent avoir également de lourds effets secondaires et par conséquent les risques et les avantages doivent être soigneusement pris en compte par les médecins et les patients. Enfin, le patient doit décider si les avantages l'emportent sur les risques avant d'accepter un traitement.
-> Bienveillance, Ethique
Notice (Règlementation)
Document présent à l'intérieur du conditionnement d'un médicament.
Dans l’UE, la notice, comme l'information sur l’emballage extérieur et/ou le conditionnement primaire (étiquetage) constitue des obligations réglementaires et doivent être enregistré avec l'AMM. La notice doit être rédigée dans un langage clair pour le patient et doit subir des tests de lisibilité. Elle contient les informations suivantes : Qu’est-ce que le médicament X et dans quel cas est-il utilisé (identification du produit médical). Ce que vous devez savoir avant de prendre/d’utiliser X (contre-indications, mises en garde et précautions d’emploi : chez les enfants et les adolescents ; avec d’autres médicaments ; avec des aliments, des boissons ou de l’alcool ; en cas de grossesse, d’allaitement, de conduite de véhicules et d’utilisation de machines ; mises en garde relatives aux excipients, le cas échéant). Comment prendre/utiliser X (posologie et mode/voies d’administration ; utilisation chez les enfants et les adolescents ; fréquence d’administration ; durée du traitement ; informations en cas de surdosage et/ou d’oubli d’une dose ; syndrome de sevrage, le cas échéant). Effets secondaires éventuels de X. Comment conserver X (conditions de conservation ; date d’expiration ; mises en garde relatives à l’utilisation du produit après la date d’expiration ; mises en garde contre des signes visibles de détérioration, le cas échéant). Contenu de l’emballage et autres informations (que contient X ; comment se présente X ; contenu de l’emballage ; forme pharmaceutique ; description physique ; formats de conditionnement ; coordonnées du détenteur de l’Autorisation de mise sur le marché (AMM) et du fabricant ; liste des représentants locaux (tous ou aucun) ; date d’approbation de la notice ; section mentionnant d’autres sources d’information, y compris le site Web associé au produit dans le cas de produits vendus sans ordonnance, le cas échéant). Les modèles d’informations sur les produits (dernière mise à jour : juin 2015 (version 9.1)) sont publiés sur le site Web de l’EMA.
-> Spécialité pharmaceutique
Notice d’information (Règlementation)
Document écrit qui doit être remis par l'investigateur à la personne dont le consentement à participer à une recherche est sollicitée résumant les informations sur celle-ci. (article L122-1 du Code de Santé Publique)
Ce document doit être approuvé par le CPP. L'information porte notamment sur le but et le déroulement de l’essai, les éventuels bénéfices et/ou risques attendus, la forme de l’étude et les traitements étudiés, etc.
-> Autonomie, Consentement
Observance (Médecine)
Respect par un sujet ou un malade des règles de prescription d’un médicament.
Le sujet ou le malade est dit "observant" s’il respecte les doses, le nombre et les horaires des prises, la durée du traitement. Terme anglais : compliance.
-> Analyse en intention de traiter, Analyse per protocole
Organisme prestataire de services (Méthodologie)
Personne physique ou morale chargée par un promoteur de mener à bien, à sa place, une ou plusieurs fonctions relatives à une recherche portant sur la personne humaine.
Ces activités sont mentionnées par écrit et le promoteur reste responsable de la conformité de la recherche aux dispositions législatives et règlementaires en vigueur et de la qualité et de l’intégrité de ses données. Terme anglais : Contract Research Organisation (CRO).
-> Promoteur, Investigateur
Participant (Méthodologie)
Une personne, volontaire sain ou malade, prenant part à un essai clinique, qu'elle reçoive un médicament expérimental ou qu'elle serve de témoin.
-> Autonomie, Inclusion, Volontaire

Perdu de vue (Méthodologie)
Participant à un essai clinique et dont l’investigateur n’a plus de nouvelles.
Plus le nombre de participants perdus de vue dans un essai est important, plus l’imprécision est grande sur les résultats de l’essai. Si cette sortie est liée à un effet lié aux traitements, les perdus de vue peuvent introduire un biais dans l'étude.
-> Biais, Puissance
Période d’exclusion (Méthodologie)
Période pendant laquelle une personne participant ou ayant participé à une recherche clinique ne peut prendre part à une autre.
Le dossier d’une recherche portant sur la personne humaine soumis au CPP et à l’ANSM détermine si nécessaire :
L’interdiction pour les sujets de participer à deux recherches simultanées.
L’interdiction pendant une période d’exclusion de participer à une autre recherche après la fin de leur participation à cette étude.
La durée de la période d’exclusion est variable selon la nature de la recherche et le traitement à l’étude. La durée proposée dans le protocole de la recherche est soumise à l’avis du CPP et de l’ANSM.
Période d'exclusivité (Règlementation)
Une période d'exclusivité est la période consécutive à l'autorisation d'un médicament durant laquelle aucun autre médicament similaire présentant les mêmes indications (usages prévus) ne peut être autorisé.
Le médicament est ainsi protégé de toute concurrence pendant la période d'exclusivité. Il peut exister plusieurs exclusivités commerciales distinctes en rapport avec des conditions désignées. La période d'exclusivité commerciale peut être prolongée de deux ans pour les médicaments ayant également satisfait aux exigences d'un plan d'investigation pédiatrique approuvé (PIP).
Petite molécule (Pharmacologie)
Familièrement, médicament issu de synthèse chimique de taille limitée, cette expression s’oppose à biomédicament.
-> Biomédicament
Pharmacien hospitalier (Règlementation)
Pharmacien responsable de la dispensation des médicaments au sein de l’hôpital.
Il est préalablement informé par le promoteur des recherches portant sur la personne humaine envisagées sur des médicaments ou des dispositifs médicaux stériles ou des préparations hospitalières. Ces produits sont détenus et dispensés par le pharmacien de l’établissement hospitalier. En cas de double insu il sera le seul à connaître la nature des traitements (médicament expérimental ou comparateur) administrés au participant.
-> Dispensation
Pharmacocinétique (PK) (Pharmacologie)
Branche de la pharmacologie qui étudie le sort du médicament dans un organisme vivant, c’est-à-dire l'effet de l'organisme sur le médicament.
La pharmacocinétique étudie en termes de quantité et de rapidité le comportement de l’organisme vis-à-vis du médicament : son absorption, son éventuelle fixation sur des protéines du sérum, sa distribution dans les différents tissus, ses possibles modifications (métabolisme) et son excrétion par l’organisme.
-> Absorption, Distribution, Métabolisme, Elimination, Pharmacodynamie
Pharmacodynamie (PD) (Pharmacologie)
Branche de la pharmacologie qui étudie l’ensemble des effets induits par un médicament sur un organisme vivant.
-> Pharmacologie, Pharmacocinétique
Pharmaco-économie (Pharmacologie)
Évaluation de l’ensemble des conséquences médico-économiques liées à l’usage d’un médicament.
-> Santé publique
Pharmaco-épidémiologie (Pharmacologie)
Etude des utilisations et des effets des médicaments à l’échelle d’une population.
Elle fournit une estimation de la probabilité des effets bénéfiques comme celle des effets indésirables d'un médicament sur une population. Elle implique une surveillance continue d'une population afin d'identifier tous effets indésirables et autres problèmes de sécurité.
-> Rapport bénéfice risque, Pharmacovigilance
Pharmacogénétique (Phamacologie)
Etude de l’influence de facteurs génétiques sur la pharmacocinétique ou la pharmacodynamique des médicaments dans un organisme.
Pharmacologie (Pharmacologie)
Etude de l’ensemble des propriétés des médicaments, de leurs caractéristiques physicochimiques à leur utilisation thérapeutique.
La pharmacologie comporte différentes branches incluant des approches in vitro, pharmacologie moléculaire, étude des récepteurs, activité et/ou toxicité sur des cellules, et des approches in vivo sur des modèles animaux et chez l’homme, la pharmacocinétique, pharmacodynamie, toxicologie, pharmacovigilance.
-> Pharmacodynamie, Pharmacocinétique, Pharmacovigilance
Pharmacologue (Médecine)
Pharmacien ou médecin spécialisé en pharmacologie.
Médecins ou pharmaciens qui étudient les mécanismes d’action de nouvelles molécules de l’échelle moléculaire (récepteurs) à celle d’un organisme entier (pharmacodynamie), la façon dont un organisme se comporte vis-à-vis de cette molécule et son devenir dans l’organisme, notamment au site thérapeutique recherché (pharmacocinétique), la tolérance ou la toxicité que ce produit peut entrainer (toxicologie), enfin ils accompagnent les cliniciens dans la déterminations des posologies et fréquences d’administration. D’autres pharmacologues s’attachent à améliorer l’utilisation de médicaments déjà existant par la mise au point de nouvelles stratégies thérapeutiques ou, individuellement, à l’ajustement individuel d’un traitement (pharmacologie clinique).
Pharmacovigilance (Pharmacologie)
Branche de la pharmacologie ayant pour objet la surveillance, l’évaluation et la prévention du risque d’effets indésirables résultant de l’utilisation des médicaments.
La pharmacovigilance se différencie de la toxicologie en ce qu'elle se réalise en situation réelle, c’est-à-dire après l’obtention de leur AMM.
-> Toxicité, Phase IV
Phase préclinique (Méthodologie)
Ensemble des investigations réalisées sur un nouveau médicament avant le début des essais chez l’homme, donc avant la phase I des essais cliniques.
-> Expérimentation animale, In silico, In vitro, In vivo
Phases I à IV de mise au point d’un médicament (Règlementation)
Phases systématisées dans la mise au point clinique d’un médicament.
Le développement d’un nouveau médicament chez l’homme est décrit en 3 phases successives à l’issue desquelles pourra être demandée l’autorisation de mise sur le marché (voir AMM) et une phase lui succédant.
- Phase I : étudie la tolérance, la pharmacocinétique et la pharmacodynamie du nouveau médicament chez l’homme. Les essais de phase I se déroulent le plus souvent chez des volontaires sains sauf quand le traitement étudié présente une toxicité trop importante pour être administré chez des volontaires sains (principe éthique de bienfaisance). C’est le cas notamment en cancérologie .
- Phase II : étudie la relation existant entre les doses du médicament et les effets thérapeutiques (bénéfiques) ou indésirables (toxiques) du nouveau médicament chez des malades dans l’indication envisagée du médicament.
- Phase III : étudie le rapport entre l’efficacité et la tolérance du nouveau médicament chez un grand nombre de patients exposés au médicament pendant des durées variables selon la pathologie et le mode de prescription futur du médicament, dans les conditions envisagées d’utilisation.
- Phase IV : est une phase de pharmacovigilance, qui se situe après introduction sur le marché et étudie la survenue d’éventuels effets indésirables, notamment rares, en condition normales d’utilisation.
La systématisation de la mise au point d’un médicament en phases successives peut être assouplie dans certaines situations. Ainsi la phase I de médicaments dotés d’une toxicité propre ou ne manifestant leur effet que chez des malades sera réalisée sur des malades. Dans les pathologies concernant un petit nombre de sujets, et donc de potentiels participants, les phases II et III peuvent être réunies (phase II-III).
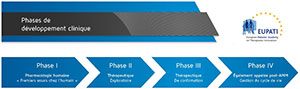
Phénotype (Biologie)
Ensemble des caractères observables d'un individu, dus aux facteurs héréditaires (génotype) ou aux modifications apportées par le milieu environnant.
-> Génotype
PK/PD (Pharmacologie)
Relations entre la dose et l'efficacité d'un médicament.
Tenant compte de phénomènes comme l'absorption et l'élimination d'un médicament, les relations PK/PD sont utilisées pour décrire et/ou prédire l'action d'un médicament en lien avec la dose et la durée d'exposition.
-> Pharmacocinétique, Pharmacodynamie
Placebo (Pharmacologie)
Substance sans effet pharmacologique démontré utilisée dans le cadre des essais cliniques comme comparateur du médicament étudié.
Le but est de distinguer l’effet pharmacologique propre d’une substance de l’effet suggestif produit par l’administration d’un produit quel qu’il soit (effet placebo), lié à la confiance qu’a le patient dans des effets de ce produit. Le Placebo a la même forme, la même couleur, la même odeur que celles du médicament étudié mais ne contient pas de substance active.
-> Effet placebo, Comparateur
Population de l'étude (Méthodologie)
Ensemble des individus participant à une étude.
Dans un essai clinique, les critères d'inclusion et d'exclusion décrivent à qui pourra ou ne pourra pas être proposé de participer, et par conséquent définissent les caractéristiques de la population de l'étude.
-> Eligibilité, Valeur externe
Population-cible (Méthodologie)
Population de patients visée par un traitement ou une intervention de santé publique.
Les essais cliniques de phase III permettent de mieux définir la population-cible d’un traitement, à laquelle il est possible d’extrapoler les résultats des essais cliniques et qui sera validée par l’AMM du médicament.
-> Santé publique, Valeur externe
Post-commercialisation (Règlementation)
Période consécutive à l'autorisation de mise sur le marché d'un médicament.
C’est pendant cette phase que peuvent être identifiés les effets secondaires rares.
-> Phase IV
Pratique clinique (Médecine)
Régime de traitement habituellement suivi pour traiter, prévenir ou diagnostiquer une maladie ou un trouble.
La pratique clinique correspond au traitement et à la gestion des patients par des professionnels de la santé sur la base de preuves cliniques. Des directives de pratique clinique ont été établies pour aider les professionnels de la santé et les patients dans les décisions sur les soins de santé appropriés dans des circonstances spécifiques.
-> Bonnes pratiques cliniques
Prédisposé (Médecine)
Une personne prédisposée à une maladie a plus de risque que les autres de la développer un jour.
Une prédisposition ne provoque pas en elle-même la maladie, mais cette dernière peut à terme être déclenchée par certains facteurs environnementaux notamment liés au mode de vie, comme le tabagisme ou l'alimentation. Les tests génétiques peuvent identifier les personnes qui sont génétiquement prédisposées à certaines maladies.
Preuve de concept (Méthodologie)
Démonstration d’une caractéristique cruciale dans l’utilisation d’une molécule comme médicament.
Les études de preuve de concept interviennent précocement dans le développement clinique d'un médicament. Les essais de phase II débutent généralement par un essai de preuve de concept, destiné à démontrer que le médicament interagit avec la cible souhaitée et est donc susceptible d’agir sur l'affection concernée.
Principes éthiques de la recherche (Ethique)
Principes philosophiques et moraux visant à protéger les participants à la recherche clinique et à garantir l'intégrité de la recherche.
Initialement portés par des textes déclaratifs (Code de Nüremberg, Rapport Belmont, Déclaration d’Helsinki, Convention d’Oviedo) ils sont aujourd’hui à la base de réglementations, soit en étant explicitement contraignantes, soit par référence aux textes déclaratifs. Ces différents textes mettent en avant trois principes éthiques majeurs : la Bienfaisance ou Non Malfaisance, l’Autonomie ou respect de la personne et la Justice distributive.
-> Code de Nüremberg, Déclaration d’Helsinki, Rapport Belmont, Déclaration d’Oviedo, Bienfaisance, Autonomie, Justice distributive
Produits biologiques à effet thérapeutique (Pharmacologie)
Produits incluant les produits de thérapie cellulaire, les produits de thérapie génique et les organes, les tissus et les cellules modifiés à des fins thérapeutiques.
Produits de thérapie cellulaire (Phamacologie)
Produits biologiques à effet thérapeutique issus de préparations de cellules vivantes humaines ou animales.
Produits de thérapie génique (Phamacologie)
Produits biologiques à effet thérapeutique visant à transférer du matériel génétique.
Promoteur (Règlementation)
Personne physique ou morale qui prend l’initiative d’une recherche portant sur la personne humaine, en assure la gestion et vérifie que son financement est prévu.
En pratique, le promoteur doit choisir l’investigateur, recruter des ARC, soumettre le protocole de la recherche à l’avis du CPP et à l’ANSM, contracter une assurance couvrant les conséquences éventuelles de cette recherche, et déclarer à l’ANSM, au CPP et aux investigateurs les événements indésirables graves inattendus survenus au cours de la recherche. De plus, il est habituel que le promoteur prenne en charge les coûts de la recherche mais ce n’est pas obligatoire puisqu’un tiers peut financer la recherche sans se porter obligatoirement promoteur. A la fin de la recherche, le promoteur avise l’autorité compétente et le CPP que la recherche est terminée, indique les raisons qui motivent l’arrêt de la recherche lorsqu’il est anticipé Terme anglais : sponsor.
-> Recherche clinique, Soumission, Investigateur
Protocole (Méthodologie)
Document rassemblant tous les éléments descriptifs d’une recherche portant sur la personne humaine et qui précise les conditions dans lesquelles cette recherche doit être réalisée et gérée.
Ce document daté, approuvé par le promoteur et par l’investigateur, est soumis au CPP et à l’ANSM, en intégrant le cas échéant, les modifications successives. Le terme « protocole» recouvre les versions successives du protocole ainsi que ses modifications.
-> Recherche clinique, Bonnes pratiques cliniques
Puissance (Statistique)
Mesure de la capacité d’une recherche à répondre à la question posée.
Dans un essai comparatif, la puissance croit avec la différence entre les groupes de traitement et l’effectif de l’étude, elle est limitée par la variabilité du critère d’évaluation. Un manque de puissance (lié à une faiblesse de différence entre groupes ou d’effectif, ou une trop grande variabilité à l’intérieur de chaque groupe) peut conduire à une étude non conclusive et donc éthiquement critiquable au regard de l’engagement des participants.
-> Valeur de p, Effectif
Qualité de vie (QdV) (Méthodologie)
Mesure du bien être initialement utilisée en économie de la santé.
Elle exprime l'effet de facteurs tels que les symptômes, la douleur, la santé psychologique et le bien-être sur la vie des gens. Les mesures de qualité de vie liée à la santé peuvent être utilisées pour calculer l'impact des traitements sur les vies des patients et à ce titre faire partie des critères d’évaluation d’une recherche clinique.
-> Critères d’évaluation
Randomisation (Méthodologie)
voir : Tirage au sort
Rapport Belmont (Ethique)
Rapport de la Commission nationale pour la protection des sujets humains dans le cadre de la recherche biomédicale et béhavioriste.
Publié en 1979 aux Etats-Unis, il pose 3 principes éthiques de base : le respect des personnes par leur consentement libre et éclairé, le calcul bénéfices/risques de la recherche et la justice en pratiquant une sélection équitable des sujets de recherche.
-> Ethique, Déclaration d’Helsinki
Rapport bénéfice/risque (Pharmacologie)
Rapport théorique qui existe entre le bénéfice thérapeutique attendu du traitement et le risque potentiel d’effets indésirables de ce traitement.
Réaction indésirable inattendue (Pharmacologie)
Réaction nocive et imprévue à un médicament qui ne concorde pas avec les informations relatives au produit ou avec les caractéristiques du médicament.
-> Toxicité, Résumé des caractéristiques du produit, Effet indésirable
Recherche clinique (Règlementation)
Recherche impliquant la personne humaine, ou portant sur des produits biologiques issus du corps humain, ou sur des données individuelles de santé.
-> Essai clinique
Recherche impliquant la personne humaine (Règlementation)
Recherches organisées et pratiquées sur l'être humain en vue du développement des connaissances biologiques ou médicales.
Il existe trois catégories de recherches impliquant la personne humaine :
1° Les recherches interventionnelles qui comportent une intervention sur la personne non justifiée par sa prise en charge habituelle ;
2° Les recherches interventionnelles qui ne comportent que des risques et des contraintes minimes, dont la liste est fixée par arrêté du ministre chargé de la santé, après avis du directeur général de l'Agence nationale de sécurité du médicament et des produits de santé ;
3° Les recherches non interventionnelles qui ne comportent aucun risque ni contrainte dans lesquelles tous les actes sont pratiqués et les produits utilisés de manière habituelle. (Article1121-1 du Code de Santé Publique).
Recrutement (Méthodologie)
Processus utilisé par les investigateurs pour sélectionner des personnes éligibles à une étude clinique.
Le recrutement est basé sur des critères d'inclusion et de non inclusion qui sont précisés dans le protocole de l'étude.
-> Inclusion, Consentement
Récurrence (Médecine)
Réapparition d'un signe, d'un symptôme ou manifestation d'une maladie après un temps pendant lequel ils n'étaient plus détectables.
Par exemple, la réapparition de cellules cancéreuses au site de la tumeur d'origine ou à distance. Le risque de récurrence dépend de différents facteurs : type de maladie, type et durée du traitement.
Référence bibliographique (Méthodologie)
Enregistrement formaté des caractéristiques d’une publication scientifique.
Il comprend : le nom des auteurs, le titre de l’article, le nom du journal, l’année de publication, le volume et les numéros de la première et de la dernière page de l’article. Toute publication scientifique possède une référence qui permet de la retrouver dans des banques de données et est requise lorsqu’elle est citée dans un autre texte.
Relation effet-dose (Pharmacologie)
Relation existant entre les doses d’un médicament administrées et les effets (bénéfiques ou indésirables) observés à la suite de cette administration.
Rentabilité (Pharmacologie)
Mesure d’efficacité d’un médicament utilisé en pharmaco-économie.
La rentabilité y est étudiée en observant les résultats de différentes interventions en mesurant un seul résultat, généralement en unités « naturelles » (par exemple, années de vie gagnées, décès évités, crises cardiaques évitées ou cas détectés). D'autres interventions sont alors comparées en termes de coût par unité (naturelle) d'efficience afin d'évaluer la manière dont la rentabilité est fournie. Cette évaluation économique aide les décideurs à déterminer où allouer des ressources de soin limitées. La rentabilité n'est qu'un critère parmi d’autres qui devraient être utilisés pour déterminer si les interventions vont être mises à disposition ou non. D'autres problèmes, tels que l'équité, les besoins et les priorités devraient également faire partie du processus de prise de décision.
-> Pharmaco-économie
Représentant légal (RL) (Règlementation)
Individu autorisé (par exemple par un tribunal) à prendre des décisions au nom d'une autre personne.
Dans des circonstances où un patient n'est pas en mesure de donner lui-même un consentement éclairé (par exemple, des patients en soins intensifs), un représentant légal peut parfois donner un consentement ou une autorisation en son nom, y compris concernant des traitements médicaux.
-> Consentement, Autonomie
Respect de la personne (Ethique)
Concept selon lequel tous les individus ont le droit d'exercer leur pleine autonomie.
Dans la recherche, ce principe vise à garantir que les souhaits d'un participant sont respectés, même s'ils sont différents de ceux du chercheur.
-> Autonomie, Ethique
Résumé des caractéristiques du produit (RCP) (Règlementation)
Document approuvé dans le cadre de l'autorisation de mise sur le marché d’un médicament en décrivant la nature et les propriétés.
Il est destiné aux professionnels de la santé et fournit les informations suivantes :
modalités d'utilisation du médicament ;
affections dans lesquelles le médicament peut être utilisé (indications thérapeutiques) ;
posologie ;
mode d'administration ;
conditions dans lesquelles le médicament ne doit pas être utilisé (contre-indications) ;
mises en garde spéciales ;
mécanisme d'action du médicament ;
et effets indésirables éventuels.
Le RCP fournit des informations plus précises que la notice du médicament.
Risque (Médecine)
Probabilité qu’un préjudice se produise lors de l’utilisation d’un traitement dans la pratique clinique (risque thérapeutique) ou dans le cadre d’une recherche (risque expérimental).
Le préjudice peut être physique, psychologique, social ou économique. Aucun essai clinique ne peut être réputé sans risque. Avant de décider de prendre part ou non, les participants doivent être conscients à la fois des bénéfices et des risques (voir consentement éclairé). Dans un essai clinique les risques peuvent avoir différentes origines : effets secondaires d’un traitement, attribution d’un médicament moins efficace que le traitement de référence par exemple. Les risques liés à des effets secondaires imprévus sont plus importants dans les premiers stades des essais cliniques.
-> Effet indésirable
Saisie (Méthodologie)
Enregistrement d’informations dans une base de données.
Dans un essai clinique les informations collectées dans les cahiers d’observation sont saisies après vérification.
-> Base de données, Cahier d’observation
Santé publique (Médecine)
Approche de la santé des individus à l’échelle d’une population sous ses aspects préventifs, curatifs et éducatifs.
Sensibilité (Méthodologie)
Capacité d'une mesure ou d'une expérimentation à détecter une différence, par exemple entre deux groupes de participants qui reçoivent des médicaments différents au cours d'un essai clinique.
-> Critère d’évaluation, Spécificité
Service médical rendu (SMR) (Règlementation)
Evaluation chiffrée de l’intérêt thérapeutique d’un nouveau médicament en vue de sa prise en charge par l’assurance maladie.
L’évaluation du SMR est réalisé par la Commission de transparence, instance scientifique au sein de la Haute Autorité de Santé (HAS), composée de médecins, pharmaciens, spécialistes en méthodologie et épidémiologie, et variable dans le temps.
Son évaluation prend en compte plusieurs aspects :
D’une part la gravité de la pathologie pour laquelle le médicament est indiqué, d’autre part l’efficacité et effets indésirables du médicament dans une indication donnée, sa place dans la stratégie thérapeutique (notamment au regard des autres thérapies disponibles) et son intérêt pour la santé publique.
Son apport comparé aux traitements existants dans la même pathologie est chiffré par un autre indicateur pharmaco-économique, l’Amélioration du Service Médical Rendu (ASMR).
Seuil « aucun effet indésirable observé » (Pharmacologie)
Dose la plus élevée d'un médicament qui a été testée et où il n'y a pas eu d'augmentation de la fréquence de tout effet indésirable (biologiquement ou statistiquement significative) par rapport à son témoin.
Cette évaluation est souvent désignée par son acronyme en anglais : NOAEL, pour No Observable Adverse Effect Level. Bien que l'approche NOAEL implique la prise en considération de certaines propriétés pharmacocinétiques et pharmacodynamiques, il se centre sur l'estimation de la dose «sûre » la plus élevée, à la recherche d'un paramètre de sécurité basé sur le seuil toxicologique.
-> Toxicité
Significativité statistique (Statistique)
Mesure de la confiance que l’on peut avoir dans la véracité d’un résultat.
Elle est exprimée par le risque d’erreur à affirmer que le résultat obtenu ne soit pas le fruit du hasard.
-> Valeur de p
Simple insu (Méthodologie)
Procédure expérimentale où le participant ne connait pas le groupe de traitement auquel il appartient alors que l’équipe de recherche en est informé.
-> Insu, Double insu, Comparateur, Placebo
Soumission (Règlementation)
Procédure administrative de demande d’enregistrement.
Pour qu'un médicament puisse être commercialisé, une soumission doit être adressée à l'autorité réglementaire compétente, à l'Agence européenne des médicaments (EMA), par exemple. Les soumissions fournissent des informations complètes sur le médicament, sa formulation, les essais qu'il a subis, ses indications ainsi que ses risques et bénéfices.
-> Autorisation de Mise sur le Marché, Agence Nationale de Sécurité du Médicament et des Produits de Santé, European Medicine Agency.
Sous-groupe (Méthodologie)
Groupe d’individus possédant des caractéristiques particulières au sein d'une population.
Ces caractéristiques peuvent concerner les individus (âge, sexe, origine, …) ou la maladie dont ils sont atteints (stade, type, échec de traitements antérieurs …). La stratification lors de l’inclusion en fonction de ces caractéristiques autorise une analyse par sous-groupes. Des sous-groupes déterminés après la réalisation d’une recherche ne permettent pas de conclure sur une efficacité différentielle, mais uniquement de générer des hypothèses qui devront être confirmées dans une autre étude.
-> Stratification
Spécialité pharmaceutique (Règlementation)
Médicament fabriqué industriellement identifié par une dénomination et par une ou plusieurs présentations. (Art. L.5111-2 du Code de la Santé Publique)
-> Médicament
Spécificité (Méthodologie)
Capacité d'une expérimentation ou d'un essai à détecter correctement et uniquement l'effet particulier étudié.
Un essai insuffisamment spécifique peut donner un résultat faussement positif.
-> Sensibilité
Stratification (Méthodologie)
Prise en compte lors de l’inclusion des participants de caractéristiques individuelles, pour que différents sous-groupes soient également représentés dans les groupes de traitement.
Cette procédure permet d’une part d’éviter un biais d’inclusion, d’autre part de réaliser ultérieurement une analyse stratifiée qui renseigne sur l’activité différentielle d’un produit, définissant éventuellement les sous-groupes qui peuvent plus utilement en bénéficier. Une analyse stratifiée non prévue dans le protocole n’a qu’une valeur indicative.
-> Sous-groupes, Biais d’inclusion
Suivi de la recherche (Méthodologie)
Surveillance du déroulement d’une recherche de sa conformité au protocole.
Il veille à ce que et que les données soient recueillies et rapportées conformément au protocole, aux bonnes pratiques cliniques et aux dispositions législatives en vigueur. Ces contrôles de qualité sont réalisés en début et en cours d’essai sous la responsabilité du promoteur de la recherche. Cette fonction et assurée par le moniteur ou l’Assistant de Recherche Clinique (ARC) recruté par le promoteur à cet effet pour la durée de la recherche portant sur la personne humaine.
-> ARC, Bonnes pratiques cliniques, Inspection
Surveillance passive (Méthodologie)
Utilisation de services de santé locaux pour recueillir des données relatives à l'incidence des maladies ou aux effets indésirables des médicaments.
Elle repose sur le personnel et sur les services, qui font partie intégrante d'un réseau chargé de la collecte de données et de la génération de rapports. Aucune recherche active n'est effectuée sur les cas. Par exemple, la surveillance passive est la méthode la plus couramment utilisée pour surveiller l'incidence des maladies évitables par la vaccination. Elle permet aux organismes nationaux et internationaux d'identifier l'apparition des foyers de maladie potentiels et d'organiser la fourniture des vaccins.
-> Pharmacovigilance
Suspension d'un essai clinique (Règlementation)
Interruption de la conduite d'un essai clinique par l’autorité compétente.
-> ANSM
Taille de l'échantillon (Méthodologie)
Dans un essai clinique, la taille de l'échantillon est le nombre de participants ou d'observations réalisées.
Il doit être suffisant pour que les différences entre les groupes dans l'essai puissent être détectées. Lors de la conception de l’étude, une estimation de la taille de l'échantillon est requise et doit être spécifiée dans le protocole d'étude. Cette estimation permet de déterminer la probabilité qu'un effet réel soit identifié comme étant statistiquement significatif. Inversement, il est injustifié qu’un médicament soit testé sur un trop grand nombre de patients.
-> Puissance, Valeur de p, Respect de la personne
Technologies « omiques » (Biologie)
Champs d'étude tels que la génomique (étude des génomes), la protéomique (étude des protéines) ou la métabolomique (étude du métabolisme).
Elles analysent de grands volumes de données car elles évaluent les génomes, protéomes ou métabolomes entiers.
-> Génomique
Tests précliniques (Méthodologie)
Avant qu’un produit expérimental soit administré à l’homme il doit subir un certain nombre de tests précliniques réalisés in vitro, sur des cellules, des tissus et/ou des animaux.
L’objectif principal des tests précliniques consiste à déterminer la sécurité d’emploi d’un médicament. Les tests précliniques étudient tous les effets nocifs du médicament sur l’organisme dus à la pharmacologie du médicament, tels que:
Effets toxiques, par exemple sur l’appareil génital.
L'éventualité de modifications génétiques induites.
L'éventualité d'un effet mutagène, c’est-à-dire cause d’une tumeur cancéreuse.
La toxicité est mesurée pour différentes doses, ou en fonction de la durée d’utilisation du médicament. La réversibilité de toute toxicité est également étudiée.
Les informations des tests précliniques sont utilisées pour la planification d’essais cliniques sur les êtres humains. Elles sont utilisées pour déterminer la dose initialement administrée. Elles suggèrent également les signes cliniques à rechercher afin de détecter des effets indésirables.

Tirage au sort (Méthodologie)
Mode de décision reposant sur le hasard.
L’attribution d’un traitement à une personne se prêtant à la recherche par tirage au sort. Elle permet de réduire les biais résultant de l’attribution d’un traitement donné aux participants présentant certaines particularités (âge, stade de la maladie, genre). Le tirage au sort concerne le traitement reçu ou l’ordre des administrations dans les études en cross-over. La procédure de tirage au sort est généralement couplée à celle d’insu, l’administration « en ouvert » d’un comparateur, notamment un placebo pouvant être psychologiquement difficile Dans le cadre de la recherche clinique, le terme anglais de randomisation est le plus couramment employé.
-> Insu, Essai en ouvert, Essai croisé
Tolérance (Pharmacologie)
Capacité de l'organisme à supporter une certaine substance.
On désigne parfois par « tolérance au médicament », la diminution de la réaction à des doses constantes répétées d'un médicament ou la nécessité d'augmenter les doses pour maintenir un effet constant. Cette « tolérance au médicament » peut entraîner une dépendance physique (physiologique) ou émotionnelle, qui est un état d'adaptation associé à un syndrome de sevrage à l'arrêt d'une exposition répétée à un médicament.
-> Toxicité, Effet indésirable
Toxicité (Pharmacologie)
Degré auquel une substance chimique ou biologique peut nuire à un organisme vivant.
Les effets néfastes peuvent concerner des organes, des tissus ou des cellules spécifiques ou l'organisme entier. Le développement des médicaments est un processus pas à pas qui implique l'évaluation de leur innocuité aussi bien pour les animaux que pour les êtres humains. Les études d'innocuité non cliniques (avant les essais sur l'homme) doivent permettre d'identifier les effets toxiques potentiels susceptibles de se produire dans les conditions de l'essai clinique ultérieur.
-> Effet indésirable
Toxicologie pour la reproduction (Pharmacologie)
Etude des effets négatifs d'un médicament qui interfère avec la fonction sexuelle normale et la fertilité.
Ces effets négatifs incluent des réactions indésirables sur la fonction sexuelle et la fertilité chez les adultes hommes et femmes, ainsi qu'une toxicité pour le développement de la descendance.
-> Effet indésirable
Traitement de référence (Méthodologie)
Traitement, reconnu par la communauté scientifique internationale et les autorités de santé comme le mieux établi scientifiquement dans une indication donnée.
Il sert de comparateur avec la méthode de soin à l’étude ; celui-ci sera alors qualifié de plus, autant ou moins efficace et/ou de mieux, autant ou moins bien toléré.
-> Comparateur, Placebo, Essai comparatif
Utilité (Pharmacologie)
Capacité (perçue) d'un élément à satisfaire des besoins ou des souhaits.
En économie de la santé, les utilités mesurent l'intensité des préférences des patients. Par exemple, l'importance de divers facteurs pour les patients, tels que les symptômes, la douleur et la santé psychologique. L'impact de nouveaux traitements sur ces facteurs, et par conséquent sur la qualité de vie (QdV), peut alors être calculé. C'est une approche courante utilisée par les entités d'évaluation des technologies de la santé qui conseillent si des traitements devraient, ou non, être financés (par exemple) par des services de santé gouvernementaux.
-> Pharmaco-économie
Valeur p (Statistique)
Mesure statistique du risque d’affirmer qu’une différence observée est liée au traitement alors qu’elle n’est que le fruit du hasard.
Le niveau de signification requis doit être établi avant le début de la collecte de données et il est généralement choisi à 5 % (ou 0,05). Cependant, d’autres niveaux peuvent être utilisés en fonction de l’étude. S’il génère une valeur p inférieure ou égale au niveau de signification, le résultat est considéré comme statistiquement significatif. Cela est généralement écrit sous la forme suivante : p≤0,05. Au contraire, si la valeur de p est grande, la différence observée est probablement un résultat hasardeux et l’idée qu’il n’existe pas de différence entre les traitements est maintenue.
Validité externe (Méthodologie)
Capacité des conclusions d’une étude à être généralisée, en particulier à la population cible.
L’évaluation de la validité externe repose sur la comparaison des caractéristiques des participants à l’étude avec celles de la population cible, et la possibilité de reproduction des conditions de réalisation du traitement expérimenté dans des circonstances de soin de routine.
-> Population cible
Validité interne (Méthodologie)
Qualité méthodologique du résultat obtenu.
Cette notion inclut la significativité statistique du résultat, le plus souvent exprimé par la valeur de p, et l’absence de biais .Les procédures de randomisation et d’insu permettent en principe de se prémunir respectivement des biais d’inclusion et d’interprétation.
-> Significativité, Valeur p, Biais
Volontaire (Ethique)
Personne qui agit sans contrainte, selon son libre arbitre.
Toute personne qui se prête à une recherche portant sur la personne humaine doit être volontaire au sens du Code de la Santé Publique ( art. L1122-1) "Préalablement à la réalisation d’une recherche portant sur la personne humaine, le consentement libre, éclairé et exprès de celle-ci doit être recueilli après que l’investigateur ait fourni les informations prévues par la loi".
-> Autonomie
Volontaire sain (Règlementation)
Personne en bonne santé qui participe de son plein gré à une recherche clinique.
Tout médicament est d’abord évalué chez le volontaire sain (cf. essai de phase I) à l’exception de médicaments aux effets indésirables lourds (Ex: oncologie) et lorsque l’éthique ne le permet pas. Dans ces cas le traitement est évalué chez des malades.
-> Autonomie
Vulnérable (Ethique)
Caractérise, dans le cadre de la recherche clinique, les personnes dont la participation, pour différentes raisons, présente un risque particulier. Elles sont, sauf exception, exclues des recherches cliniques.
Les participants ou populations vulnérables sont des individus ou des groupes d'individus incapables de donner leur consentement éclairé à participer à un essai clinique, tels que les enfants ou les personnes affectées par des problèmes de santé mentale, ou encore qui risquent de subir des pressions pour les inciter à prendre part à l'étude. Il peut également s'agir de personnes dont la volonté de participer à un essai clinique peut être indûment influencée par ce qu'elles attendent de leur participation. Si un essai doit inclure des personnes issues de populations vulnérables, il faudra veiller tout particulièrement à protéger leur bien-être, tant de la part des investigateurs que du comité d'éthique qui révise le protocole de l'essai.
"Ne peuvent être sollicitées pour des recherches que si des recherches d'une efficacité comparable ne peuvent être effectuées sur une autre catégorie de la population et dans les conditions suivantes :
- soit l'importance du bénéfice escompté pour ces personnes est de nature à justifier le risque prévisible encouru ;
- soit ces recherches se justifient au regard du bénéfice escompté pour d'autres personnes placées dans la même situation. Dans ce cas, les risques prévisibles et les contraintes que comporte la recherche doivent présenter un caractère minimal". (articles: 1121 5 à 9 du Code de Santé Publique)











